McCune-Albright Syndrome is a rare, heterogenous disorder caused by a sporadic, somatic, post-zygotic mutation in GNAS. It is most commonly manifested as the triad of polyostotic fibrous dysplasia, café-au-lait hyperpigmentation along with hyperfunctional endocrinopathies.2,3 The syndrome has a female predilection1 with an estimated prevalence between 1/100,000 and 1/1,000,000.4 The most easily visually assessed feature is café-au-lait skin pigmentation characterized by hyperpigmented maculae with pathognonomic irregular and serpinginous borders classically described as the "Coast of Maine," which are mainly found on the anterior and posterior neck, gluteal region, thorax, back, shoulder and pelvis.2 The most common feature of the syndrome, fibrous dysplasia (FD) which can be monoostotic (single bone involvement) or polyostotic (multiple bone involvement) is characterized by the replacement of medullary bone with a poorly organized fibrous tissue containing trabeculae of immature bone.2,6 The third component of the classical triad is hyperfunctioning endocrinopathies which may include precocious puberty, hyperthyroidism, pituitaryadenomas, primary adrenal hyperplasia, hypophosphatemia and ovarian cysts.5 Gonadotropin independent precocious puberty is the most common endocrinopathy which is more common in girls. Other endocrine and non-endocrine organs like the heart, adrenal, thyroid, pituitary and liver can be involved.1,2
Rarely, manifestations of rickets and osteomalacia are present in children and adolescents diagnosed to have fibrous dysplasia. These patients have the characteristic biochemical profile of hypophosphatemia with phosphaturia, attributed to the hypersecretion of a phosphatonin fibroblast growth factor 23 (FGF23) from dysplastic bone lesions.1,2,4
We present a case of the classic triad of MAS along with hypophosphatemic rickets. Only a few cases have been reported with simultaneous involvement of precocious puberty and hypophosphatemic rickets along with fibrous dysplasia and café-au-lait macules.
A 3½ year-old girl was admitted for evaluation of enlargement of both breasts along with vaginal bleeding for 23½ months. The girl was born of a non-consanguineous marriage, full-term, normal vaginal institutional delivery with cephalic presentation and cried immediately after birth. There were no feeding difficulties, no jaundice, no lactose intolerance and no developmental delay. There were no other chronic illnesses, no decreased appetite, galactorrhea, hypothyroidism, hypocortisolism, head trauma, meningitis, seizures, encephalitis, visual field defect, brain surgery, radiation, defective smell, difficulty in vision, difficulty in walking or deformity of extremities. There was no significant maternal drug intake.
AnthropometryThe following is her anthropometric data: Height: 90 cms (3-10 percentile), Arm Span: 88 cms, Mean Parental Height: 161.5 cm, Weight: 15 kg (50-75 percentile).
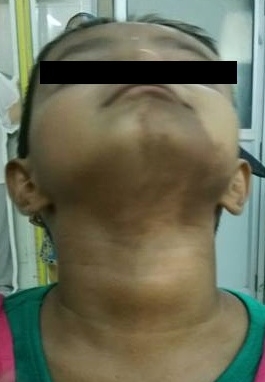
Physical ExaminationHer physical exam revealed a conscious, fully oriented female child, with feminine facies and body contour, antalgic gait, thyroid gland not enlarged, normal hair distribution, Marshall & Tanner stage: A0 P1 B3, There was no hirsutism, nor clitoromegaly. Her pulse was 70/min, regular, with normal peripheral pulses; BP was 90/52 mm Hg, with no postural fall. Chest, cardiovascular, abdominal, and neurologic examinations were normal.There was tenderness over the left thigh with restriction in movement of the left hip joint.There were café-au-lait spots on the left side of the face. [Figure 1]
Click here to download Figure 1Figure 1. Café-au-lait spot involving left side of chin and upper neck.
Laboratory Investigations
Basal levels of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) were low (0.4 and 0.7 IU/L, respectively), with no change in response to stimulation by 100 mg of triptorelin (Table 1).
Click here to download Table 1Table 1.Basic biochemical and hormonal profile.
Pelvic ultrasound revealed left ovarian cysts measuring 3.70 & 3.4 cm. The uterus was enlarged with a length of 4.6 cm (normal = 3 cm) and had a prominent endometrium.
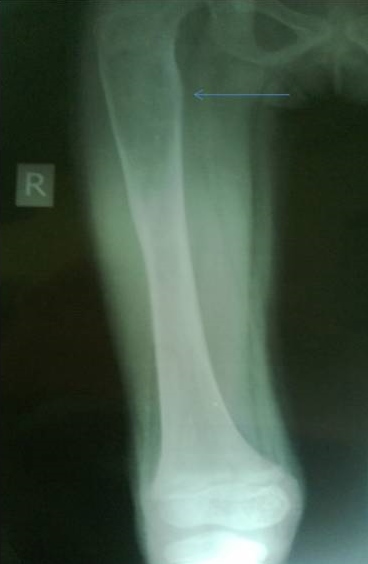
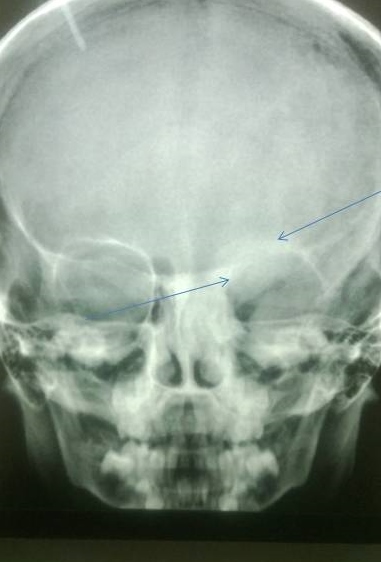
X-ray of the lower limbs showed fibrous dysplasia involving the left femur [Figure 2] and skull x-ray showed ground glass appearance of the left orbit. [Figure 3]
Click here to download Figure 2Figure 2.Well defined geographic lesions showing ground glass haze and sclerotic margins is seen involving left femoral metaphysics and diaphysis.
Click here to download Figure 3
Figure 3. Lesions showing ground glass haze are seen involving roof, lateral and medial walls of left orbit
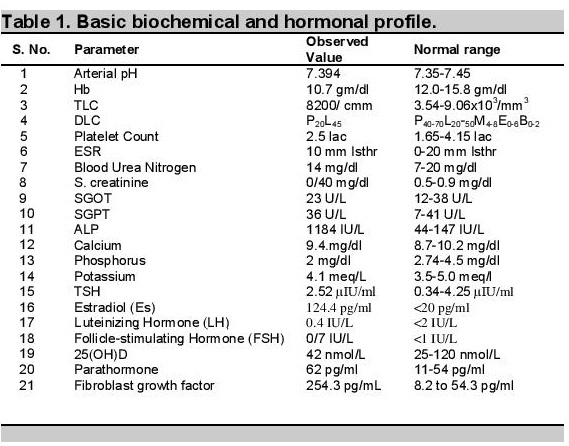
Other investigations revealed normal serum calcium (9.4 mg/dL), phosphorus was reduced to 2 mg/dL (normal 2.7-4.5 mg/dL), markedly elevated serum alkaline phosphatase 1184 IU/L (normal 145-420 U/L), normal potassium (4.1 meq/L), normal 25(OH),D level of 42 nmol/L (25-120 nmol/L). Parathormone level was mildly elevated at 62 pg/ml (normal 11 - 54 pg/ml), fibroblast growth factor (FGF-23): 254.3 RU/mL.
Arterial blood pH was 7.394 and urine pH was 5.3. Urine examination for glucose and amino acids was negative ruling out the possibility of proximal renal tubular acidosis.
After evaluation, she was diagnosed with McCune-Albright syndrome with hypophosphatemic rickets, and was started on letrazole along with phosphate supplement and calcitriol.
McCune Albright Syndrome is a rare, multisystem disorder with female predilection, first described separately by Donovan McCune and Fuller Albright in 1937, in a group of children presenting with café-au-lait maculae, bone deformities and hyperfunctioning endocrinopathies attributed to an activating mutation of Gs gene.1 The syndrome is caused by a postzygotic somatic mutation in GNAS1 gene located on 20q13-13.29, encoding for the alpha subunit of the stimulatory G protein, resulting in the constitutive activity of the gene products. Normally, on binding of ligands to the receptor, the Gsα gets stimulated and dissociates from the receptor to activate adenylyl cyclase enzyme to increase the production of cAMP which mediates further signaling cascade. On inactivation, Gsα again reattaches with receptor for ligand mediated reactivation.1,2
The clinical presentation is heterogeneous with involvement of endocrine & non-endocrine organs depending on the number and the types of cells carrying the GNAS1 mutation.2 Similarly, the syndrome has varied evolution and progression. Usually the syndrome is diagnosed on the basis of the classic triad of fibrous dysplasia which develops due to the mutation in osteoprogenitor cells without differentiation, hyperfunctioning endocrinopathies along with isosexual precocious puberty and café-au-lait maculae mostly developing between the age of 4 months to 2 years but can be present at birth.2,8
Fibrous dysplasia usually manifests during the first decade of life as aching pain, pathological fractures, limb asymmetry and deformities and is usually polyostotic, commonly involving the long bones, ribs and skull. Involvement varies greatly in severity from small, asymptomatic areas only detected on bone scan to highly disfiguring lesions leading to the pathological fractures and impingement of vital organs.7
Sometimes MAS is associated rarely with hypophosphatemic rickets, a group of disorders with the common denominator of defective renal phosphate reabsorption leading to phosphaturia and hypophosphatemia. The most common form is X-linked hypophosphatemic rickets. Acquired forms of hypophosphatemic rickets are associated with mesenchymal tumors, hemangiomas and soft-tissue tumors.1,2 Hypophosphatemic rickets associated with MAS is caused by the overproduction of the phosphatonin FGF23 by the cells in the dysplastic region of the bone.
McCune-Albright syndrome is considered a complex disease because of its varied manifestations and severity. No definite treatment and prenatal diagnosis is possible at present. A recent novel technique of PCR for activating mutation in the peripheral blood cells can help diagnose the disease.2,7,10 Depending on the number and extent of the endocrine and non-endocrine manifestations, various medical and surgical therapies can be offered.9
Among all the manifestations, the management of fibrous dysplasia is particularly difficult especially in children. Bisphosphonates like pamidronate, have a good short term safety profile and relieve pain, prevent fractures and partially resolve lesions. Sometimes corrective surgeries can be offered for fibrous dysplasia lesions.11
Precocious puberty can also be an early manifestation of McCune-Albright syndrome and the etiologic diagnosis of early sexual precocity is based on careful history and physical examination.
Children with precocious puberty should be evaluated for endocrinopathies and hormonal studies may be necessary in such cases.
1. Albright F, Butler AM, Hampton AO. Syndrome characterized by osteitisfibrosadisseminata, areas of pigmentation and endocrine dysfunction, with precocious puberty in females. N Engl J Med. 1937;216(17):727-46. http://dx.doi.org/10.1056/NEJM193704292161701.
2.Lumbroso S, Paris F, Sultan C. Activating Gsα mutations: Analysis of 113 patients with signs of McCune-Albright syndrome - A European Collaborative Study. J Clin Endocrinol Metab. 2004;89(5):2107-2113. http://dx.doi.org/10.1210/jc.2003-031225.
3. Chopra R, Chander A, Jacob JJ. The eye as a window to rare endocrine disorders. Indian J Endocr Metab. 2012;16(3):331-8. http://dx.doi.org/10.4103/2230-8210.95659.
4. Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis. 2008; 3:12. http://dx.doi.org/10.1186/1750-1172-3-12.
5. Xavier SP, Ribeiro MC, Sicchieri LG, Brentegani LG, Lacerda SA. Clinical, microscopic and imaging findings associated to McCune-Albright syndrome: Report of two cases. Braz Dent J. 2008;19(2):165-70. http://dx.doi.org/10.1590/s0103-64402008000200014 .
6. Medow JE, Agrawal BM, Resnick DK. Polyostotic fibrous dysplasia of the cervical spine: Case report and review of the literature. Spine J. 2007;7(6):712-5. http://dx.doi/10.1016/j.spinee.2006.10.023.
7. DiCaprio MR, Enneking WF. Fibrous dysplasia.Pathophysiology, evaluation, and treatment.J Bone Joint Surg Am. 2005; 87(8):1848-64. http://dx.doi.org/10.2106/JBJS.D.02942.
8.Papadopoulou M, Doula S, Kitsios K, Kaltsas T, Kosta K. A boy with McCune-Albright syndrome associated with GH secreting pituitary microadenoma. Clinical findings and response to treatment. Hormones (Athens). 2006;5(3):205-9. http://dx.doi.org/10.14310/horm.2002.11186.
9. Bhadada SK, Bhansali A, Das S, Singh R, Sen R, Agarwal A, et al. Fibrous dysplasia and McCune-Albright syndrome: An experience from a tertiary care centre in north India. Indian J Med Res. 2011;133(5):504-9.
10. Lietman SA, Ding C, Levine MA. A highly sensitive polymerase chain reaction method detects activating mutations of the GNAS gene in peripheral blood cells in McCune-Albright syndrome or isolated fibrous dysplasia. J Bone Joint Surg Am. 2005; 87(11):2489-94. http://dx.doi.org/10.2106/JBJS.E.00160.
11. Lane JM, Khan SN, O'Connor WJ, Nydick M, Hommen JP, Schneider R, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res. 2001; 382:6-12.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.