Periodic paralysis is one of the dramatic and life-threatening complications of Graves' disease (GD). Muscular weakness in GD can be due to thyrotoxic myopathy, periodic paralysis and associated myasthenia. Thyrotoxic periodic paralysis (TPP) is characterized by the acute onset of severe hypokalemia and profound proximal muscle weakness in patients with thyrotoxicosis.1 The neuromuscular symptoms of TPP are quite similar to that of hypokalemic periodic paralysis except for the presence of signs and symptoms of thyrotoxicosis. Early clinical recognition is pivotal as management strategies of both conditions are different. Antithyroid drugs (ATD) and beta blockers are cornerstones of therapy. Potassium replacement has a risk of rebound hyperkalemia, potassium correction should precede with caution and frequent monitoring. Achieving and maintaining a euthyroid state is key, as it avoids the recurrence of paralytic attack.
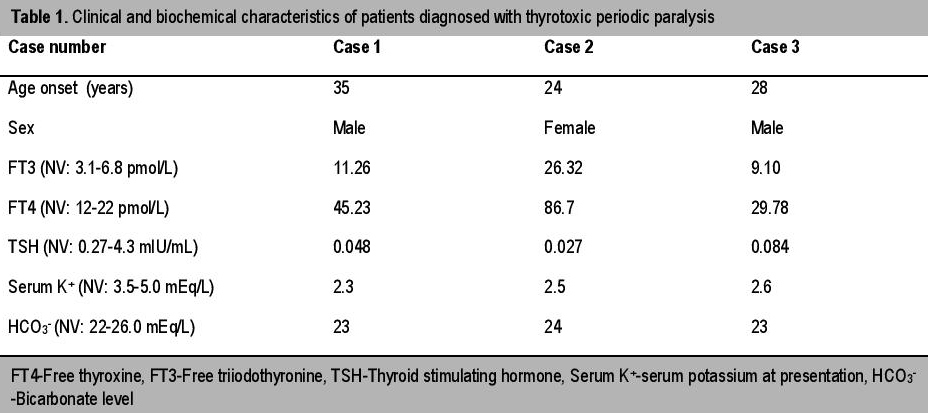
A 35-year-old man presented to the emergency department with complaint of generalized weakness of all four limbs. No precipitating factors could be identified on enquiry.There was history of similar attacks, occuring thrice in the past six months but it resolved on its own within a few hours and he was never hospitalized. The weakness appeared at night time. The patient had no history of fever, diarrheal episode, palpitation, nervousness or weight loss. There was no history of similar attack in his family members. On examination, the patient was conscious, oriented and afebrile. The pulse rate was 104/minute and blood pressure was 130/80 mm Hg. Neck examination revealed a small, diffuse, non-tender, grade 1 goiter without any bruit. He had no cranial nerve deficit. The motor power was grade 2/5 in all four limbs and deep tendon reflexes (DTRs) were slightly depressed. There was no sensory or bladder involvement. No thyroid-associated ophthalmopathy or dermopathy was detected. Electrocardiogram (ECG) revealed sinus tachycardia and presence of U waves in leads v2 to v4. Important biochemical investigations and thyroid function tests are summarized in Table 1. Renal function tests, liver function tests and glycemic status were normal. Based on above findings, a diagnosis of TPP was made. He was given intravenous potassium solution (potassium chloride), beta blockers (propranolol) and ATD (methimazole) (Table 2). He made an uneventful recovery from paralysis within six hours. Subsequently, nerve conduction study (NCS) and electromyography (EMG) were done which revealed no abnormality. Ultrasonography (USG) of the thyroid gland revealed diffuse enlarged thyroid gland with increased vascularity. Technetium 99m scan (Tc 99 m scan) was done later on and it showed a hyperfunctioning gland with increased diffuse tracer uptake. During a nine-month follow-up period, the patient was euthyroid with anti-thyroid medications and was free from any paralytic attack.
Click here to download Table 1Table 1. Clinical and biochemical characteristics of patients diagnosed with thyrotoxic periodic paralysis
Click here to download Table 1
Table 2. . Summary of treatment received and time taken for recovery from paralysis among patients
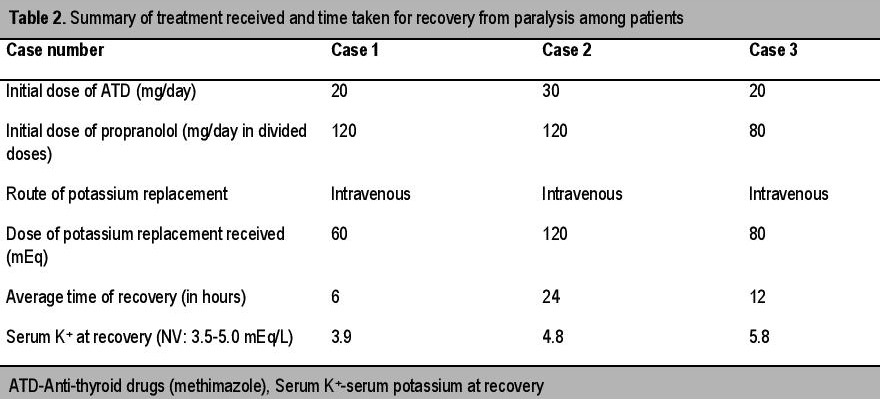
A 24-year-old female presented with sudden onset of weakness of both lower limbs for four hours. There was no history of a similar attack in the past or in any family members. The patient was diagnosed with thyrotoxicosis two years prior and was prescribed ATD. In her last clinical follow up she was prescribed 15 mg of methimazole. However, she was non-compliant to medications. Prior to presentation, she has had symptoms of thyrotoxicosis for one year. She was conscious and afebrile. She had a pulse rate of 120/minute and blood pressure was 140/76 mm Hg. Neck examination revealed a diffuse, non-tender goiter without any bruit. She had normal cortical functions and there was no cranial nerve deficit. Motor strength was grade 3/5 in both lower limbs with grade 4/5 in upper limbs. DTRs were diminished. There was no sensory or bladder involvement. There was no associated ophthalmopathy or dermopathy. Biochemical evaluation confirmed presence of thyrotoxicosis and hypokalemia (Table 1). The rest of the parameters were normal. An USG (including color doppler) of the thyroid showed diffusely enlarged goiter with marked increase in intraparenchymal blood flow. In view of frank signs of hyperthyroidism, the diagnosis of TPP was straightforward. This case elucidates the need to explore the possibility of TPP in females even though it is much more common in males. She was managed successfully with potassium supplementation, ATD and beta blockers (Table 2). However, recovery from weakness and hypokalemia took a longer time than the previous case (24 hours). She was counseled regarding compliance to ATD and need for Iodine 131 ablation in case of failure to achieve remission with ATD. She was well till last follow up with the medication and without report of any recurrence.
A 28-year-old man presented with quadriparesis for the last three hours. There was no history of a previous attack. The patient gave a history of strenuous exercise earlier in the day. He also complained of generalized muscle pain prior to paralysis. The patient had no history of fever, diarrheal episode, palpitations, nervousness or weight loss. There was no history of similar attack in any family member. On evaluation, the pulse rate was 96/minute and blood pressure was 126/84 mm Hg. Neck examination revealed a small goiter without any bruit. He had normal cortical functions with no cranial nerve deficit. The power was grade 2/5 in all four limbs and DTRs were depressed. There was no sensory or bladder involvement. There was no associated ophthalmopathy or dermopathy. ECG revealed presence of U waves in leads v2 to v5. Biochemical investigation revealed hypokalemia (Table 1). Renal function tests, liver function tests and glycemic status were normal. Routine thyroid function tests (TFT) revealed elevated thyroid hormone levels (Table 1). Thyrotoxicosis was solely detected due to routine screening (to rule out possible association of hyperthyroidism in such cases of hypokalemic periodic paralysis). This illustrates that TPP should be regarded a strong differential for periodic paralysis especially in males, despite the absence of overt signs of thyrotoxicosis. Basing on above findings, a diagnosis of TPP was made. After receiving treatment (Table 2) he made an uneventful recovery. During his replacement(started with 10 meq/hr), his serum potassium reached 5.8 mEq/L with no untoward clinical sequelae. Hence, caution should be exerted during potassium replacement to prevent the phenomenon of rebound hyperkalemia and its dangerous side effects. Subsequent nerve conduction study (NCS) and electromyography (EMG) revealed no abnormality. Technetium 99 m scan (Tc 99 m scan) revealed a hyperfunctioning gland with diffuse tracer uptake compatible with diagnosis of GD. The patient was maintained well with ATD without any attack of TPP.
The association of periodic paralysis and thyrotoxicosis had been recognized as early as 1902 by Rosenfeld.2 The first description of the entity in English literature was made by Dunlap and Kepler in 1931, who described four such patients.3 In Asian populations (where the incidence of TPP is highest), it usually appears between the ages of 20 and 40 years of life, coinciding with the peak age for hyperthyroidism.4 The male to female ratio ranges from 17:1 to 70:1 despite the fact that hyperthyroidism is more common in females (female to male ratio of 9:1).5 The attack is characterized by recurrent, transient episodes of muscle weakness that range from mild weakness to complete flaccid paralysis. Neurologic examination during an attack demonstrates weakness, usually affecting proximal more than distal muscles and the legs more than the arms. Sensory system and bladder involvement are never seen.6 Diminished DTRs are typical of TPP in contrast to hyperreflexia seen in the thyrotoxic state. Most patients with TPP have only mildly elevated serum thyroid hormone levels and only about 10% of patients may have mild thyrotoxic symptoms.5 Rare, life-threatening complications of TPP include bulbar palsy requiring mechanical ventilation and fatal arrhythmias including ventricular fibrillation and ventricular tachycardia.6
Although the majority of cases of thyrotoxicosis associated with TPP are due to Graves' disease, TPP can appear with thyrotoxicosis of any origin. Patients with TPP have been reported with thyroiditis, toxic adenoma or toxic nodular goiter.6 The precipitating factors of TPP reported in the literature include high carbohydrate ingestion, strenuous exercise, trauma, acute upper respiratory tract infection, high-salt diet, emotional stress, exposure to cold, alcohol ingestion, menstruation and use of drugs.6-8 TPP may be preceded by prodromal symptoms such as muscle pain, cramps or stiffness of muscles of the affected limbs.6 TPP does not usually recur once the patient is euthyroid. Achieving proper control of hyperthyroidism is essential. Hsieh et al reported that before achieving the euthyroid status, the rate of recurrent attacks was as high as 62.2%, peaking in the first 3 months after TPP diagnosis.8
Hypokalemia occurs due to a massive shift of potassium into the cells rather than net loss from the body. Excess thyroid hormone may predispose to paralytic episodes by increasing the susceptibility to epinephrine or insulin, and therefore increasing Na+/K+-ATPase activity in the beta-adrenergic receptors in skeletal muscles, which leads to potassium shift into the cells.9 The hypokalemia observed in these cases is due to the increased K+ influx into a cell secondary to the increase in the activity of the Na+/K+-ATPase pump and by the hyperinsulinemic response to carbohydrate intake in patients susceptible to TPP.10,11 Androgens can also increase the activity of the Na+/K+-ATPase pump, which explains the higher incidence of the disease in young males.11 Recent studies have shown that susceptibility to TPP can be conferred by loss-of-function mutations in the skeletal muscle specific Kir channel, Kir2.6 and loci on 17q involved in Kir2.1 gene expression.12-17 These findings also suggest that reduced basal muscular Kir current may also be an important mechanism of TPP.9,11 The dual effects of increased intracellular K,sup>+ influx from activated Na+/K+-ATPase and decreased K+ efflux from defective Kir channels potentiate the serum hypokalemia that upsets membrane polarization and muscle excitability in TPP.9-11
Studies exploring the genetic susceptibility for TPP indicate that defects of the skeletal muscle-specific inward rectifying K+ (Kir) channel, Kir2.6, encoded by the KCNJ18 gene, is associated with a proportion of TPP patients mainly from the United States, Brazil, France and Singapore.12 Other important developments are that gene polymorphisms (rs623011and rs312691) at 17q24.3 may affect the expression of KCNJ2 gene (encoding Kir2.1) in Hong Kong and Thai populations.14,15 The presence of different HLA antigen subtypes such as DRw8, A2, Bw22, Aw19, B17, B5, and Bw46 in certain ethnic populations (Singaporean, Chinese and Japanese) may make them more susceptible to TPP.6,7 Hence, TPP is considered as an endocrine channelopathy with genetic background.11
In a recently published, large study by Li et al, the authors have explored the genetic susceptibility of Chinese patients with TPP. They concluded that KCNJ18 gene mutations occurred in a small proportion (3.1%) of these patients with TPP.18 The patients with KCNJ18 mutation had significant clinical difference than patients without KCNJ18 mutation. The former group had shorter attack duration, higher prevalence of muscle soreness and weakness recurrence than the latter. The loci polymorphisms (rs623011and rs312691) at 17q24.3 are significant risk factors for TPP.18
Laboratory findings include hypokalemia, normal bicarbonate level, hypophosphatemia, mild hypomagnesemia and normal blood pH. Urinary findings include low potassium excretion rate (low urinary potassium/creatinine ratio), hypercalciuria and hypophosphaturia. Abnormal thyroid function tests (low TSH; elevated free and total T4 and T3) are hallmarks of the disease. Electrocardiographic findings may be characteristic of hypokalemia, with increased P-wave amplitude, prolonged PR interval, widened QRS complexes, decreased T-wave amplitude, and U waves. Unlike hypokalemia from other causes, sinus tachycardia predominates in patients with TPP. Other electrocardiographic abnormalities include atrioventricular block, atrial fibrillation, ventricular fibrillation and asystole.19,20 Differential diagnoses of TPP include the causes of periodic hypokalemia, e.g., familial hypokalemic periodic paralysis, sporadic periodic paralysis and other causes of potassium wasting disorders (diuretics use, primary hyperaldosteronism, Bartter's syndrome, Gitelman's syndrome, renal tubular acidosis) causing marked hypokalemia. Hence, urinary potassium excretion measurement and arterial blood gas analysis are essential requisites to rule out such disorders.
ManagementTPP is managed by correction of hypokalemia and treatment of the underlying hyperthyroid state. It is achieved by intravenous or oral potassium replacement to hasten muscle recovery and prevent cardiopulmonary complications. It has been shown that adequate control of hyperthyroidism by any one of the following modalities, that is, use of antithyroid drugs and non-selective beta blockers, radioactive iodine or surgical thyroidectomy are successful in preventing TPP. Nonspecific beta-adrenergic blockers like propranolol have also been proposed as an alternative treatment to ameliorate the paralysis without rebound hyperkalemia and raise the serum levels of potassium and phosphate. High-dose oral propranolol (3-4mg/kg orally) alone has been reported to rapidly abort the paralysis.21
Propranolol, but not the selective beta1-blocker metoprolol, also effectively prevented recurrence of paralytic attacks or inhibited paralysis induced by a carbohydrate load. At a dose of 40 mg four times a day, propranolol prevented paralysis in carbohydrate-induced TPP in about two thirds of cases by inhibiting the activity of Na+/K+-ATPase.6,22
In a retrospective case series, patients who received intravenous potassium recovered more quickly than those who received oral supplementation.23 There may be a delayed response of a few hours following potassium administration.24 Rebound hyperkalemia occurred in approximately 40% of patients with TPP, especially if they received more than 90 mEq of potassium chloride within the first 24 hours.6 There is a positive correlation between the dose of potassium chloride administered and the degree of rebound hyperkalemia.4,6,25 This is because hypokalemia in TPP is due to redistribution of potassium rather than net loss from body. Lower doses of potassium chloride may be effective while lowering the patient's risk of hyperkalemia.25 Required doses of potassium supplementation are variable and range from 10 to 200 mEq.6 Lower doses of potassium chloride may be effective while lowering the patient's risk of hyperkalemia. Thus potassium supplementation should be given at a slow rate unless there are cardiopulmonary complications.6 There is no definite role of potassium supplementation for prophylaxis against further paralytic attacks and should be avoided.6 Therefore, monitoring the serum potassium levels during the treatment and suspending the infusion at the first sign of the muscular force recovery is recommended.13
In a recent study by Chang et al, the authors evaluated the efficacy of various treatment modalities for managing patients of GD complicated by TPP. They found out that patients with GD who had TPP appeared to have a greater chance of thyrotoxic and paralysis relapse when managed by ATD alone.26 A more definitive treatment modality such as surgery or radioiodine therapy is more prudent for patients with GD with TPP. When radioiodine is chosen over surgery, a higher dose (>550 MBq) should be employed in order to cure hyperthyroidism.26 In two recent studies, the TPP relapse rate during withdrawal or tapering of ATD stood at 29.6% and 50% respectively.17,26 The discrepancy in relapse rates in these studies may be due to difference in study population and follow-up period.
The possibility of TPP should always be borne in mind while dealing with a case of periodic paralysis. Though the prevalence of TPP is highest among Asians, due to migration of population, several cases have been also reported from western countries. As signs of hyperthyroidism are often subtle, it is advisable to routinely measure thyroid hormone levels in high risk population presenting with hypokalemic paralysis. It helps to secure the diagnosis and initiate proper therapy. The patients should be promptly treated with potassium supplements and nonselective beta-blockers to prevent life-threatening complications of hypokalemia. The correction of hypokalemia in TPP is a tricky clinical situation as potassium replacement carries a risk of rebound hyperkalemia. Achieving euthyroidism is the key in preventing recurrence of disease. Physicians should be aware of this life threatening and atypical presentation of Graves' disease.
1. McFadzean AJ, Yeung R. Periodic paralysis complicating thyrotoxicosis in Chinese. Br Med J. 1967;1:451-455. http://dx.doi.org/10.1136/bmj.1.5538.451.
2. Rosenfeld M. Akute aufsteigende Lahmungen bei Morbus Basedow. Berl Klin Wochenschr. 1902;39:538-40.
3. Dunlap HF, Kepler EJ. A syndrome resembling familial periodic paralysis occurring in the course of exolphthalmic goiter. Endocrinology. 1931;15:541-6.
4. Manoukian MA, Foote JA, Crapo LM. Clinical and metabolic features of thyrotoxic periodic paralysis in 24 episodes. Arch Intern Med. 1999;159(6):601-606. http://dx.doi.org/10.1001/archinte.159.6.601.
5. Ko GTC, Chow CC, Yeung VTF, Chan HHL, Li JKY, Cockram CS. Thyrotoxic periodic paralysis in a Chinese population. Q J Med. 1996;89:463-468. http://dx.doi.org/10.1093/qjmed/89.6.463.
6. Kung AW. Clinical review: Thyrotoxic periodic paralysis: A diagnostic challenge. J Clin Endocrinol Metab.2006;91(7):2490-5. http://dx.doi.org/10.1210/jc.2006-0356.
7. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80(1):99-105. http://dx.doi.org/10.4065/80.1.99.
8. Hsieh MJ, Lyu RK, Chang WN, et al. Hypokalemic thyrotoxic periodic paralysis: Clinical characteristics and predictors of recurrent paralytic attacks. Eur J Neurol. 2008; 15(6):559-564. http://dx.doi.org/10.1111/j.1468-1331.2008.02132.x.
9. van Dam GM, Reisman Y, van Wieringen K. Hypokalaemic thyrotoxic periodic paralysis: Case report and review of an Oriental syndrome. Neth J Med. 1996; 49(2):90-97. http://dx.doi.org/10.1016/0300-2977(96)00014-9.
10. Lee KO, Taylor EA, Oh VMS, Cheah JS, Aw SE. Hyperinsulinaemia in thyrotoxic hypokalaemic periodic paralysis. Lancet. 1991;337(8749):1063-1064. http:/dx.doi.org/10.1016/0140-6736(91)91710-C.
11. Lin SH, Huang CL. Mechanism of thyrotoxic periodic paralysis. J Am Soc Nephrol. 2012; 23: 985-8.
12. Ryan DP, da Silva MR, Soong TW, Fontaine B, Donaldson MR, Kung AWC, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140(1):88-98. http://dx.doi.org/10.1016/j.cell.2009.12.024.
13. Maciel RM, Lindsey SC and Dias da Silva MR. Novel etiopathophysiological aspects of thyrotoxic periodic paralysis. Nat Rev Endocrinol. 2011;7:657-667. http://dx.doi.org/10.1038/nrendo.2011.58.
14. Cheung CL, Lau KS, Ho AY, Lee KK, Tiu SC, Lau EY, et al. Genome-wide association study identifies a susceptibility locus for thyrotoxic periodic paralysis at 17q24.3. Nature Genetics. 2012;44:1026-1029. http://dx.doi.org/10.1038/ng.2367.
15. Jongjaroenprasert W, Phusantisampan T, Mahasirimongkol S, Mushiroda T, Hirankarn N, Snabboon T, et al. A genome-wide association study identifies novel susceptibility genetic variation for thyrotoxic hypokalemic periodic paralysis. J Hum Genet. 2012;57: 301-304. http://dx.doi.org/10.1038/jhg.2012.20.
16. Chu PY, Cheng CJ, Tseng MH, Yang SS, Chen HC, Lin SH. Genetic variant rs623011 (17q24.3) associates with non-familial thyrotoxic and sporadic hypokalemic paralysis. Clin Chim Acta. 2012;414:105-108. http://dx.doi.org/10.1016/j.cca.2012.08.004.
17. Chang CC, Cheng CJ, Sung CC, Chiueh TS, Lee CH, Chau T, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: Focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-36. http://dx.doi.org/10.1530/EJE-13-0381.
18. Li X, Yao S, Xiang Y, et al. The clinical and genetic features in a cohort of mainland Chinese patients with thyrotoxic periodic paralysis. BMC Neurology. 2015;15:38. http://dx.doi.org/10.1186/s12883-015-0290-8.
19. Boccalandro C, Lopez L, Boccalandro F, Lavis V. Electrocardiographic changes in thyrotoxic periodic paralysis. Am J Cardiol. 2003; 91(6):775-777. http://dx.doi.org/10.1016/S0002-9149(02)03431-8.
20. Hsu Y, Lin Y, Chau T, Liou JT, Kuo SW, Lin SH. Electrocardiographic manifestations in patients with thyrotoxic periodic paralysis. Am J Med Sci. 2003; 326: 128-32.
21. Lin SH, Lin YF. Propranolol rapidly reverses paralysis, hypokalemia and hypophosphatemia in thyrotoxic periodic paralysis. Am J Kidney Dis. 2001;37(3):620-623. http://dx.doi.org/10.1053/ajkd.2001.22090.
22. Yeung RT, Tse TF. Thyrotoxic periodic paralysis: Effect of propranolol. Am J Med. 1974; 57: 584-90.
23. Cesur M, Bayram F, Temel MA, et al. Thyrotoxic hypokalaemic periodic paralysis in a Turkish population: Three new case reports and analysis of the case series. Clin Endocrinol (Oxf) 2008;68(1):143-152. http://dx.doi.org/10.1111/j.1365-2265.2007.03014.x.
24. Tassone H, Moulin A, Henderson SO. The pitfalls of potassium replacement in thyrotoxic periodic paralysis: A case report and review of the literature. J Emerg Med. 2004;26(2):157-161. http://dx.doi.org/10.1016/j.emermed.2003.05.004.
25. Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544-7. http://dx.doi.org/10.1016/j.ajem.2004.09.016.
26. Chang RY, Lang BH, Chan AC, Wong KP. Evaluating the efficacy of primary treatment for Graves' disease complicated by thyrotoxic periodic paralysis. Int J Endocrinol. 2014; 2014. Article ID 949068. http://dx.doi.org/10.1155/2014/949068.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.