Glucagonoma is a rare, slow-growing alpha-cell tumor of the pancreatic islets of Langerhans.1 It is associated with systemic clinical manifestations, referred to as the "Glucagonoma Syndrome" characterized by necrolytic migratory erythema (NME), weight loss, diabetes mellitus, anemia, glossitis, cheilitis, steatorrhea, diarrhea, venous thrombosis and neuropsychiatric disturbances.2 The incidence of glucagonoma syndrome in the general population is one in 20 million. These clinical findings in association with hyperglucagonemia and pancreatic tumor establish the diagnosis.
A 43-year-old Filipino woman was admitted in 2014 due to progressive weight loss. In 2001, she was diagnosed with major depressive disorder and 3 years later, in 2004, she started to experience recurrent epigastric pain. Ultrasound of the whole abdomen showed sub-centimeter liver nodules.
Due to the persistence of recurrent abdominal pain, she underwent gastroscopy in 2005 which showed chronic gastritis. Lipase and amylase were slightly elevated.
Magnetic Resonance Cholangio Pancreatography (MRCP) showed unremarkable pancreas and bile ducts with a 0.7 cm focus in hepatic segment II, probably a hemangioma. Tumor markers for liver carcinoma were normal.
From 2006-2011, there was recurrent abdominal pain, now accompanied by loss of appetite and a 20 pound-weight loss over a span of 6 months. Whole abdominal CT-scan done every 6 months revealed stable liver nodule size.
In 2011, she was admitted for fever. Work up showed normochromic normocytic anemia with normal white blood cells (WBC), pneumonia and a nodular opacity on the left lung. Chest CT scan showed multiple pulmonary nodules and nodular pleural thickening in the left lower hemithorax. Consideration was a pleural neoplasm (mesothelioma) with pulmonary metastasis. CT scan guided biopsy of the left pleural-based mass was suspicious for granulomatous process. Acid-Fast Bacilli (AFB) smear and Mycobacterium Tuberculosis (MTB) culture of the biopsy specimens were negative. Blood culture was positive for pansensitive MTB.
CT scan of the whole abdomen showed multiple liver nodules at segment II of the liver (2.4 x 4.8 cm and 2.4 x 4.1 cm) and a hypodense nodule in the body of the pancreas (0.9 x 0.6 cm). Liver function tests were normal except for hypoalbuminemia. Tumor markers for liver and pancreatic carcinoma, autoimmune panel, and hepatitis profile were negative. She refused to have a biopsy of the liver and pancreatic mass and opted to have them monitored until the completion of TB treatment. She was maintained on anti TB medications for 18 months and gained some weight (10 lbs) after treatment.
She was diagnosed with Type 2 DM in 2012 and was maintained on pioglitazone. A dynamic liver CT scan done after TB treatment showed no significant changes of the liver nodules' size. However, the hypodense nodule in the body of pancreas was not demonstrated on repeat CT scan. CXR after TB treatment revealed regression of the patchy and fibronodular opacities in the left lung.
In 2013, she started to have recurrence of weight loss accompanied with on and off diarrhea. She also developed seborrhoic dermititis-like skin lesions on the centro-facial area and annular rash with crusted borders on the buttocks. The skin lesion was biopsied which was consistent with Necrolytic Migratory Erythema.
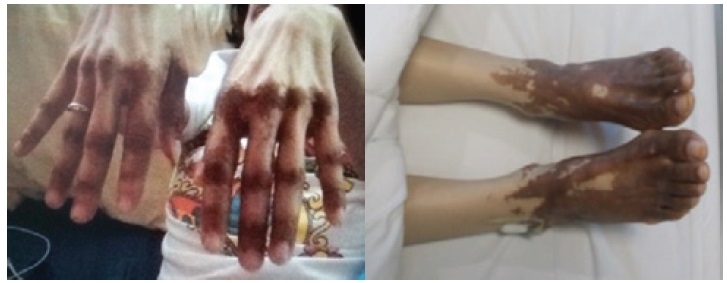
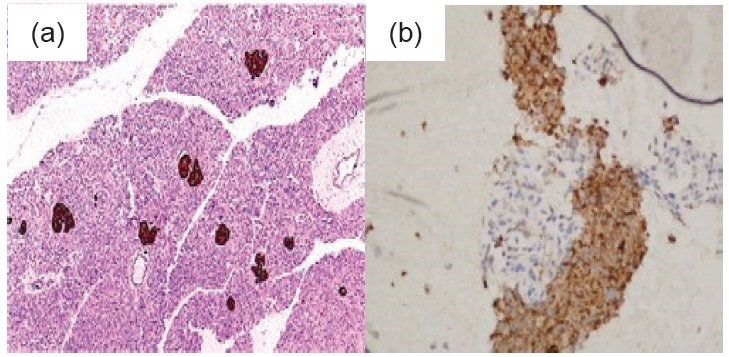
On April 2014, she was admitted for nutritional build up because at this time she was already cachectic with BMI 12.55 kg/m2. There were hyperpigmented plaques with velvety surface on the dorsum of the feet, hands, elbows (Figure 1). A repeat whole abdomen CT scan showed an increase in the size of the enhancing left lobe hepatic mass (6.6 x 6.5 x 5.6 cm). There were also hypodensities in the pancreatic body and tail (0.7 x 1.3 cm and 0.7 x 0.7 cm). Percutaneous FNAB of the pancreatic mass was consistent with pancreatic neuro endocrine neoplasm while the liver mass showed metastatic pancreatic neuro endocrine neoplasm. Immunohistochemical studies were positive for chromogranin, synaptophysin and CEA stain with a Ki67 index of 5% in the pancreas and <5% in the liver (Figure 2a and 2b). Her serum Chromogranin A was elevated at 674.579 ng/mL (NV 6.0-40.0 ng/ml). Glucagon level was also elevated 1243 pg/mL (NV <80 pg/mL). Based on all of these factors, the diagnosis is most likely Glucagonoma. She was given everolimus 5 mg/tab 1 tab once daily and ocreotide LAR 20 mg SQ then monthly thereafter.
Click here to download Figure 1Figure 1.Hyperpigmented plaques with velvety surface on dorsum of both hands and feet (photographs taken before treatment).
Click here to download Figure 2
Figure 2.Immunohistochemical stain (Chromogranin H&E, x 100) A. Islands of Langerhans B. Liver Mass.
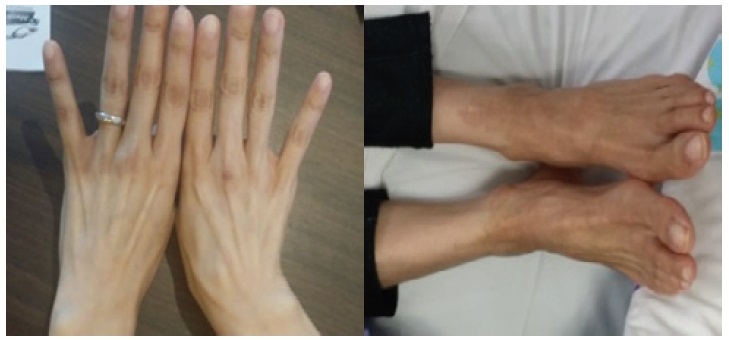
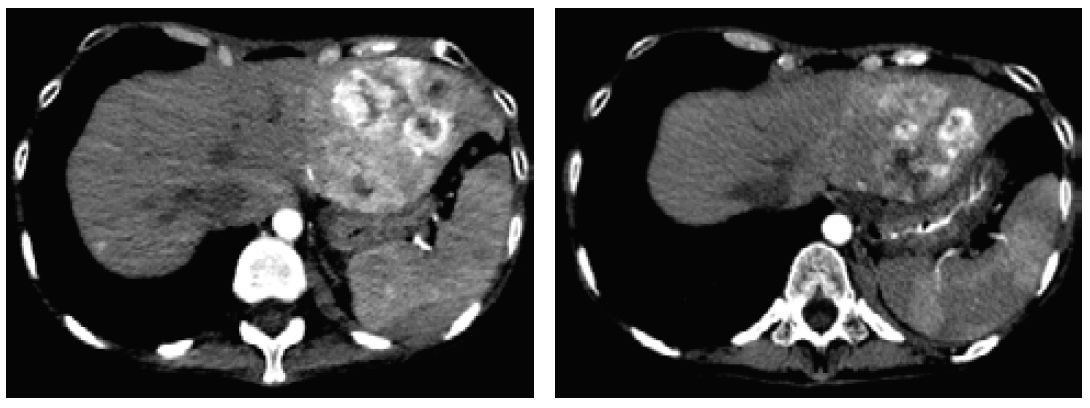
After 3 months of medical therapy, she gained 4.5 kg with improvement of the Necrolytic Migratory Erythema (Figure 3). On repeat CT scan, there was a decrease in the size of the inhomogenous mass in the left hepatic lobe (4.4 x 5.8 x 5.2 cm from 7.6 x 7.1 x 6.6 cm) (Figure 4).
Click here to download Figure 3Figure 3. Improvement of Necrolytic Migratory Erythema after treatment compared with Figure 1.
Click here to download Figure 4
Figure 4. Decreased size of the inhomogenous mass in the left hepatic lobe.
The clinical findings of weight loss, diarrhea, diabetes, and NME accompanied by elevated glucagon level more than 1000 pg/ml, satisfied the clinical and biochemical parameters for the diagnosis of Glucagonoma. Glucagonomas are rare slow-growing tumors arising from the pancreatic alpha-cells.1 Majority of cases are sporadic, however, between 5 and 17% are associated with Multiple Endocrine Neoplasia Type I (MEN 1) or, rarely, familial adenomatous polyposis. Patients with the sporadic disease present in their fifth decade of life, while those associated with MEN 1 usually have younger disease onset, with a family history of pituitary, pancreatic islet cell, or parathyroid tumors. The characteristic syndrome of glucagonoma is a result of excessive secretion of glucagon and other peptides.3
The tail of the pancreas is the most common site of glucagonoma, seen in 47% to 75% of cases, with an average size of 6 cm.4 These tumors are generally solid, circumscribed and well vascularized. By the time of diagnosis, 50% to 100% of patients already present with metastatic disease, with the liver as the most common site of metastasis, followed by regional lymph nodes, bone, adrenal gland, kidney, and lung.5 In this case, the tumors were located at the body and tail of the pancreas with liver metastasis. However, it had a different presentation that made it interesting and difficult to diagnose. In contrast with the large size of the liver metastatic lesion (6.6 x 6.5 x 5.6 cm), the sizes of the pancreatic lesions as the main tumor were very small (0.7 x 1.3 cm and 0.7 x 0.7 cm). Furthermore, the metastatic lesions in the liver appeared before the pancreatic lesion. Despite its unusual presentation, the histopathology and immunohisto-chemical studies of the pancreatic mass proved that it was a pancreatic neuroendocrine neoplasm.
Manifestations associated with glucagonoma include weight loss, NME, diabetes, cheilosis or stomatitis, and diarrhea.3,6-8 Other findings are anemia, neuropsychiatric symptoms and venous thrombosis. Weight loss, diarrhea, diabetes, and skin lesions consistent with NME were present in this patient. Many of the symptoms of glucagonoma discussed are nonspecific. As a result, glucagonoma is often diagnosed relatively late in the course of the disease. NME is often the clue which leads to the correct diagnosis.3 Even though it is not pathognomonic for glucagonoma, the presence of NME should prompt further work-up for a pancreatic neuroendocrine tumor.7 Among all the clinical manifestations, NME is the most prevalent symptoms, occurring in approximately 65 to 80 percent of patients by the time of diagnosis.3 In a retrospective study by Kindmark et al, records from 340 patients with endocrine pancreatic tumors were reassessed and 23 patients had elevated plasma glucagon levels. This was only 7% of all the endocrine pancreatic tumors in this tertiary center. Only 22% of these patients had developed diabetes prior to the diagnosis of glucagonoma. Necrolytic migratory erythema was diagnosed or clinically suspected in 52% of patients. Seventy eight percent had metastatic disease to the liver at diagnosis.9
The diagnosis is based on clinical suspicion and the demonstration of raised glucagon levels in the presence of a pancreatic tumor. Diagnostic plasma glucagon concentrations have not been precisely established but levels in excess of 1000 pg/ml can be considered biochemical evidence for glucagonoma.3
The aim of treatment should be curative whenever possible but in the majority of the cases, it is often palliative.10 Surgery remains the treatment of choice and it is the only approach that can achieve a cure. It may also still be considered even if the tumor is metastatic.11 However, if the disease is progressive or in patients in whom surgery is contraindicated, symptoms related to specific hormonal production are currently best managed with somatostatin analogues. The only proven hormonal management of NETs is by administration of somatostatin analogues. Somatostatin receptors are present in the vast majority of NETs and somatostatin analogues bind principally to the SSTR subtypes 2 (with high affinity) and 5 (with lower affinity) thus inhibiting the release of various peptide hormones. They also antagonize growth factor effects on tumor cells and at high doses, may induce apoptosis. The two commercially available somatostatin analogues in Philippines are ocretotide and lanreotide.
Two molecularly targeted agents, sunitinib and everolimus, have been approved for treatment for neuroendocrine tumors. Sunitinib is a tyrosine kinase inhibitor that acts downstream from key drivers of tumor angiogenesis, including vascular endothelial growth factor types 2 and 3, platelet-derived growth factor and stem cell factor. It showed a response rate of 72.3%.11 The mammalian target of rapamycin (mTOR) is a serine-threonine kinase, which is a major regulator of protein synthesis and stimulates cell growth, proliferation, and angiogenesis. Everolimus is an mTOR inhibitor that blocks the mTOR signaling pathway that is activated in many tumors. It showed a response rate of 77.4%.11 For this patient, surgery was not done because of poor nutritional status and the presence of liver metastasis. Hence, ocreotide LAR 20 mg SQ monthly and everolimus 5 mg daily were given.
Majority of patients with glucagonoma also suffer from a prolonged catabolic state, hence, nutritional support is an important component of therapy. Necrolytic migratory erythema may respond to somatostatin infusion that may suggest a direct effect of ocreotide.3 However, resolution usually occurs after resection of tumor.
Response to therapy includes symptomatic, hormonal and tumor responses. Tumor responses based on WHO criteria may be classified as: 1) Complete response with complete regression of all clinical, hormonal and radiological evidence of tumor; 2) Partial response with a 50% or greater reduction of all measurable tumor with hormonal and symptomatic improvement; 3) Stable disease wherein there is less than 50% reduction, or no greater than 25% increase of tumor size; 4) Progression of disease wherein there is appearance of new lesions, or an increase of 25% or more of tumor size, and hormonal and symptomatic deterioration.3
Follow-up comprises clinical, biochemical and radiological evaluation. The role of follow-up imaging and frequency depends on clinical circumstances and tumor grade. Initially, follow-up imaging may be taken at 3-6 months intervals. If the disease is relatively slow growing, the interval can be increased to 9-12 months.11 In terms of biochemical assessment, serum glucagon may be measured three and six months post-resection. Chromogranin A may be used but it should be interpreted with caution in patients on somatostatin analogues because the reduction in the chromogranin A levels may be a reflection of the effect of the somatostatin analogues rather than a reduction in the tumor size.10 For this patient, glucagon level was not repeated post therapy. However, the Chromogranin A level one-year post treatment was significantly decreased (from 674.579 ng/mL to 59.261 ng/mL).
Length of survival is directly related to both the extent of the disease at the time of diagnosis and the degree of differentiation of the tumor. Those with metastases have a 5-year and 10-year survival of 32% and 15% respectively. Age at diagnosis and stage are strongly associated with survival. Higher grade also predicted worse survival.12 Furthermore, Ki-67 is a cell proliferation marker that has some utility in predicting prognosis in neuroendocrine tumors. Five-year disease survival for low, moderate and high risk Ki-67 staining is 92%, 75%, 52% by AJCC stratification with a 10 year disease survival of 80%, 70% and 52%.13 For this patient, the Ki67 index was 5% (moderate risk) thus predicting a 5 and 10 year disease survival of 75% and 70% respectively.
In summary, we have reported a rare case of metastatic glucagonoma, with weight loss accompanied by NME as the main clinical presentations. Skin manifestations as NME are essential for early diagnosis of glucagonoma and may prevent metastatic disease.
1. Frankton S, Bloom SR. Gastrointestinal endocrine tumours. Glucagonomas. Baillières Clin Gastroenterol. 1996;10(4):697-705. http://dx.doi.org/10.1016/S0950-3528(96)90019-6.
2. Wermers RA, Fatourechi V, Wynne AG, Kvols LK, Llyod RV. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine (Baltimore). 1996;75(2):53-63. 3. Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004; 25(3):458-511. http://dx.doi.org/10.1210/er.2003-0014. 4. Shi W, Liao W, Mei X, Xiao Q, Zeng Y, Zhou Q. Necrolytic migratory erythema associated with glucagonoma syndrome. J Clin Oncol. 2010;28(20):e329-e331. http://dx.doi./org/10.1200/JCO.2009.25.7113. 5. Strosberg J, Gardner N, Kvols L. Survival and prognostic factor analysis in patients with metastatic pancreatic endocrine carcinomas. Pancreas. 2009;38(3):255-8. http://dx.doi.org/10.1097/MPA. 0b013e3181917e4e. 6. Vinik AI, Woltering EA, Warner RR, et al. NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas. 2010;39(6):713-34. http;//dx.doi.org/10.1097/MPA.0b013e3131ebaffd. 7. Van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: A clinical review. Eur J Endocrinol. 2004;151:531-37. http://dx.doi.org/10.1530/eje.0.1510531. 8. Economopoulos P, Christopoulos C. Glucagonoma. Ann Gastroenterol. 2001; 14(2):99-108. 9. Kindmark H, Sundin A, Granberg D, et al. Endocrine panrceatic tumors with glucagon hypersecretion: A retrospective study of 23 cases during 20 years. Med Oncol. 2007; 24(3):330-7. http://dx.doi.org/10.1007/s12032-007-0011-2. 10. Kulke MH, et al. NANETS treatment guidelines: Well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas. 2010;39(6):735-752. http://dx.doi.org/10.1097/MPA.0b013e3181ebb168. 11. Ramage JK, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumour (NETs)s. Gut. 2012; 61(1):6-32. http://dx.doi.org/10.1136/gutjnl-2011-300831. 12. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008; 19(10):1727-33. http://dx.doi.org/10.1093/annonc/mdn351. 13. Hamilton NA, Liu TC, Cavatiao A, Mawad K, Chen L, et al. Ki-67 predicts disease recurrence and poor prognosis in pancreatic neuroendocrine neoplasms. Surgery. 2012;152(1):107-113. http://dx.doi.org/10.1016/j.surg.2012.02.011.Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.