Neuroendocrine tumors (NET) are also known as Gastro-entero-pancreatic neuroendocrine tumors (GEP-NETs).They arise from cells of diffuse neuroendocrine systems such as enterochromaffin cells which possess secretory granules and release neurohormones.1 Pancreatic NETs present with typical syndromes which include insulinoma, gastrinoma, glucagonoma, VIPomas, somatostatinomas and non-syndromic pancreatic NETs.1
A glucagonoma is a rare pancreatic NET with an estimated incidence of 1 in 20 million people per year.2 Glucagonomas arises from islet alpha cells commonly at the tail of the pancreas.3 The World Health Organization (WHO) classification of endocrine tumors4 recommended that glucagonomas should be differentiated from non-functioning pancreatic alpha cell tumors and thus, is defined by its clinical syndromes. Glucagonomas may arise sporadically or in patients with Multiple Endocrine Neoplasia (MEN) type 15 which is rare (<3%). As depicted in our patient's case, patients with glucagonoma usually presents in the middle age group with median age of 53.5 years.6
A 58-year-old Malay female, with a background history of diabetes mellitus and hypertension for 2 years, presented with chronic, on-and-off skin lesions over the lower limbs and abdomen for the past 2 years. It usually starts with a skin blister which later ruptures and ulcerates with a necrotic patch. Other symptoms included lethargy and profound weight loss (20 kg in a year). The patient also had a history of anemia, abdominal pain and dyspepsia. Upper gastrointestinal endoscopy done previously only revealed mild gastritis. There was no history of chronic diarrhea, anorexia, history of mood changes or any history to suggest venous thrombosis.
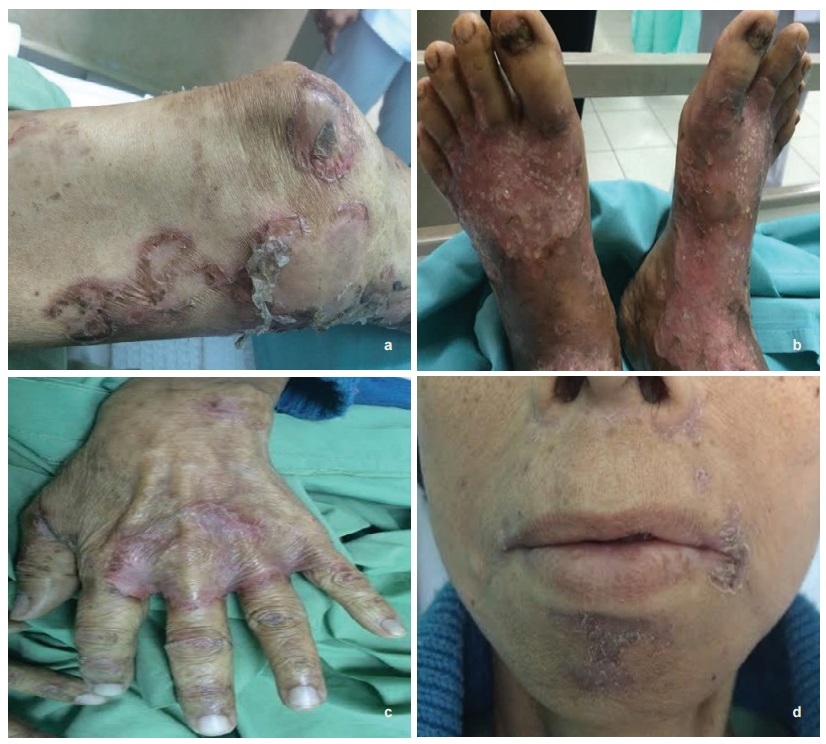
Physical examination revealed a pale, cachectic lady with erythematous, erosive scaling with crusted skin patches over the upper and lower extremities, gluteal region and perioral region (Figure 1a-d). Exudates covered certain erosive areas indicative of secondary infection. There was no angular stomatitis.
Click here to download Figure 1 (a-d).Figure 1 (a-d). Erosive scaling and crusted skin patches over the (a) thigh, (b) with exudates over both feet, (c) hand and (d) perioral area.
Investigations revealed normochromic normocytic anemia (hemoglobin of 11.3 g/dl), normal ionized serum calcium of 1.152 mmol/L (0.80-1.29) and serum phosphate ( 0.90 mmol/L) with FBS of 15 mmo/L. Hepatitis screening was negative.
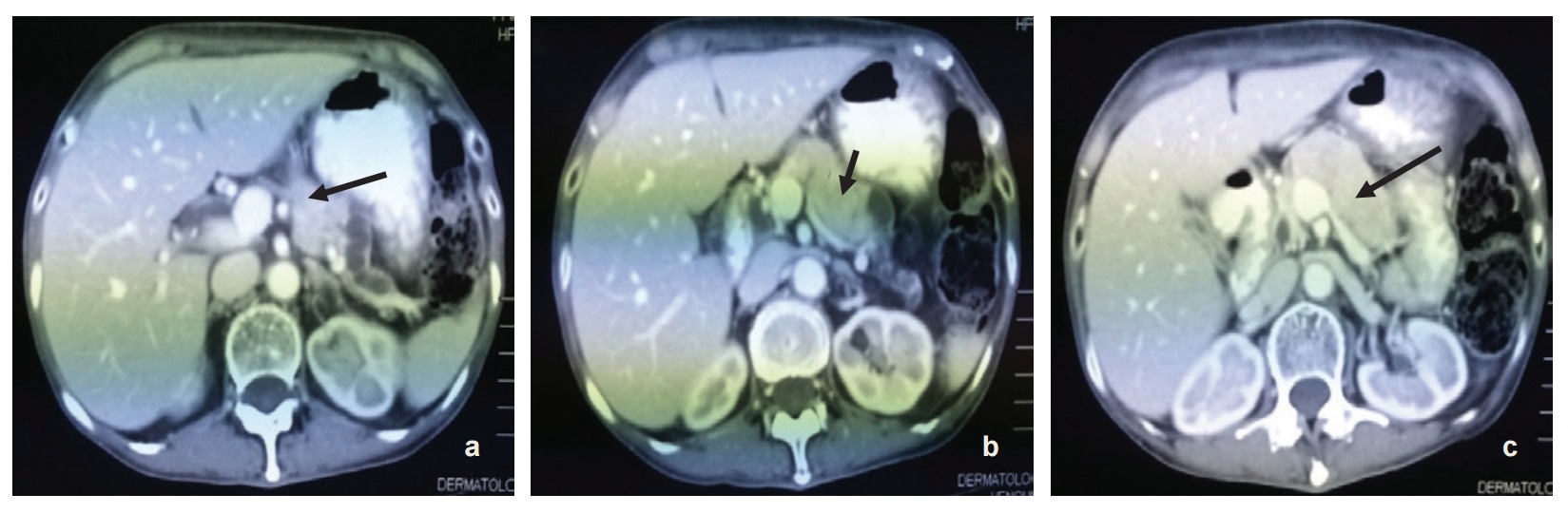
The patient was referred to the dermatology service and skin biopsy was done which revealed mild acanthosis with marked spongiosis. There was hyperkeratosis and parakeratosis as well as plasma, neutrophils and bacterial colonies on the keratin layers. The dermis shows perivascular and interstitial infiltration by lymphocytes, neutrophils and eosinophils. This findings are suggestive of infection and features are compatible with NME. Subsequently, CT scan of the abdomen was done which revealed a pancreatic neck and body tumor most likely representing glucagonoma (5.5 x 2 x 3 cm) with possible liver metastases. (Figure 2a-c). Further investigations revealed raised serum Chromogranin A >770 ng/ml (27-94) and serum glucagon of 1068 pg/ml(50-150).
Click here to download Figure 2 (a-c).Figure 2 (a-c). Radiologic findings - (serial axial view) showing tumor at the neck and body of the pancreas measuring 5.5x2x3 cm.
She was started on subcutaneous somatostatin for 2 weeks then shifted to intramuscular Sandostatin LAR 20 mcg every 4 weeks. She tolerated the medication well without any complications. Skin lesions completely resolved by 2 months with improved general well-being and weight gain. She then underwent distal pancreatectomy with wedge resection of liver nodules. Histopathological examination of the tumor revealed NET of the pancreas with liver metastases, histological grade 2. Immunohistochemical staining was strong and diffusely positive for synaptophysin and focally positive for Chromogranin A. There were no postoperative complications and patient was scheduled for surveillance CT scan of the abdomen 6 month post-operation.
Glucagonoma Syndromes (GS) are paraneoplastic syndromes consisting of a clinical triad which includes glucagon-secreting pancreatic tumor, diabetes mellitus (DM) and NME.7 Stacpoole suggested a set of diagnostic criteria for glucagonoma in which all criteria should be fulfilled.8 The diagnostic criteria consist of demonstration of the tumor by direct visualisation or radiographically, proof that the tumor shows a preponderance of glucagon-containing cells on appropriate staining and/or proof of increased tissue levels of immunoreactive glucagon; elevation of basal circulating immunoreactive glucagon; and at least one of the following coincidental findings; (a) skin rash, (b) glucose intolerance, or (c) hypoaminoacidemia. Other clinical syndromes associated with glucagonoma are stomatitis, anaemia,9 venous thromboembolism10 neuropsychiatric disturbances (most often depression and/or psychosis),11 diarrhoea and, rarely, dilated cardiomyopathy. GS can be further classified into 3 clinical types i.e., glucagonoma with skin syndrome (NME), glucagonoma with mild diabetes and glucagonoma with multiple syndromes.12 Our patient fulfilled all the diagnostic criteria for glucagonoma and belongs to the last group with presence of diabetes and NME.
NME is present in almost 70% of patients.13,14 NME is characterised by irregular annular eruptions (central healing area) with serpiginous advancing borders, erosion, and crusting, resulting in a scalded appearance.15 It is suggestive of glucagonoma but is not pathognomonic for the syndrome. NME was also reported to be found in other disorders such as hepatic dysfunction and cirrhosis,16 jejunal and rectal adenocarcinoma, villous atrophy of the small intestines and myelodysplastic syndromes.6 The skin eruption usually involves the inguinal canal, perineum, buttocks and lower extremities.7 It is characterized by cyclical pattern of spontaneous remission and exacerbation.7 The underlying cause for NME skin changes is still unclear. Increased glucagon per se does not cause NME since it was also reported in other disorders not related to high glucagon level as mentioned above. However, normalisation of glucagon level by means of surgery or somatostatin analogues invariably results in rapid resolution of NME.15 Hyperglucagonemia probably provokes the metabolic deficiency of zinc, essential fatty acids and amino acids and has been considered as a possible cause of NME. However, not all patients with NME present with these metabolic changes, and not all patients with NME and metabolic deficiency achieve complete resolution of the skin lesion with supplementation.17 Early recognition of NME is essential as it will prevent catabolic clinical features and reduce the risk of metastasis.18
Impaired fasting glycaemia or diabetes mellitus is present in 80% of patients with GS.19 The tumor secretes high levels of glucagon which in turn promotes glycogenolysis, gluconeogenesis, ketogenesis and lipidioresis by activating phosphorylase in the liver, stimulating secretion of insulin and inhibiting external secretion of the pancreas, thus resulting in hyperglycaemia.18 Hyperglucagonemia reduces plasma amino acid concentration and stimulates amino acid catabolism.18
The diagnosis of glucagonoma in this case was confirmed by elevated plasma glucagon levels in keeping with similar findings in all patients in a case series in which the glucagon level ranged from 200 to >10,000 pmol.6 Plasma glucagon levels are usually elevated 10 to 20-fold in patients with glucagonoma (normal <50 pg/mL). Concentrations above 1000 pg/mL are virtually diagnostic of glucagonoma.20 Chromogranin A (Cg A) level was elevated in our case and in half of subjects in the case series.6 Cg A is a NETs biomarker and is expressed in almost 90% of NETs independent of its site of origin.21 It can aid in the diagnosis of NETs and can also be used as prognosis and as a biomarker to monitor disease progression.21
Another biochemical result noted was anemia. Plurihormonal secretion of the tumor (serum glucagon, gastrin and chromogranin A) confers a guarded prognosis with short survival possibly due to the less-differentiated nature of the plurihormonal subtype.6 Somatostatin receptor scan (SRS) was positive in most of the patients in a case series.6 It is frequently used as a complementary method to conventional imaging like CT scan and MRI for supervision of somatostatin expression and dissemination of tumor metastases.22,23
As illustrated in our patient case, the pancreatic tumor was readily visualised by CT scan imaging and the size was comparable with the average size of 6 cm reported in a case series.24 However, the position of the tumor at the neck and body of the pancreas was not classic for glucagonoma, which is more commonly found at the tail of the pancreas (45-75%).12 CT scan is usually the initial non-invasive radiographic test. Other imaging techniques that can be used to detect the tumor are abdominal ultrasound, MRI and selective visceral angiography.7 Selective angiography of the celiac and superior mesenteric15 arteries is the gold standard in the diagnosis and localisation of glucagonoma as this tumor shows significant hypervascularity.7 In addition, this technique is able to demonstrate hepatic metastases even in a patient with a negative liver scan.7 The role of pancreatic venous sampling for glucagon levels in the diagnosis of small tumor has been reported.8 As a result of subtle and non-specific manifestations, glucagonoma is usually diagnosed late with metastatic spread on diagnosis6 and in this patient's case, liver metastases. The liver is the most common site for metastasis. Most of the time, the metastases are multiple, involving many hepatic segments and are variable in size.6 Other common sites for metastases are regional lymph nodes, bone, adrenal gland, kidney and lung.25,26
The prognosis of glucagonoma varies depending on the stage in which the tumor was diagnosed. The tumor is resistant to chemotherapy as discussed later. This tumor however is slow-growing, and prolonged survival is possible (>20 years).15 In a case with metastasis, the cause of death is unrelated to the tumor.27
Our patient was given a multimodal treatment combining medical and surgical therapy. Surgery rendered 2 patients with prolonged progression-free survival (4-6 years) and 1 patient with free disease with additional hepatic metastasectomy in a case series.6 Surgical resection can lead to prolonged disease-free remission.28-30 As for the liver metastases, control by metastasectomy, cryoablation, radiofrequency ablation, or chemoembolization has been reported.19,22
Our patient was also treated with somatostatin analogue (SSA), a potent inhibitor of glucagon secretion preoperatively and she responded well in terms of weight gain, general well-being and resolution of paraneoplastic manifestation i.e., NME. She was treated with long-acting octreotide (Sandostatin LAR), with a half-life of 3-4 weeks. As shown by the PROMID study, Octreotide LAR offers symptomatic control as well as reduction of tumor growth progression.31 However, we did not assess biochemical and imaging response of this SSA. In a case series, some response was noted following SSA therapy - 4/6 patients showed improvement of cutaneous symptoms, weight loss and general well-being, 50% reduction of plasma glucagon level in 2/6 patients but no objective tumor response on imaging.6 Other alternatives available are lanreotide autogel injection and interferon-alpha alone or in combination with SSA. New targeted therapies include antiangiogenic agents, peptide receptor radiotherapy (PRRT), mTOR inhibitors and VEGF inhibitors.1 Another available treatment modality utilised in a case series is an amino acid infusion which induced rapid long term resolution of NME rash & glossitis in 2/4 patients and local therapy to the liver (symptomatic response for external radiation to the liver and no response for trans-arterial chemoembolization or TACE).6 Intermittent infusions of amino acids and fatty acids have been associated with long-term resolution of necrolytic migratory erythema (NME).32 However, the evidence to support their use is limited to observational studies. In addition, amino acid infusions do not cause regression of tumor growth or other symptoms.32
The following are recommendations for follow-up after treatment of a pancreatic neuroendocrine tumor:33
- Three and six months postresection - history and physical examination, serum glucagon, and computed tomography/magnetic resonance imaging.
- Long-term - history and physical examination with tumor markers every 6 to 12 months for years 1 to 3, and as clinically indicated thereafter. Imaging studies are recommended only as clinically indicated.
Glucagonoma is a rare NET with a subtle, variable presentation leading to late diagnosis. Therefore increased awareness and recognition of Glucagonoma Syndrome is vital in order for early diagnosis before development of metastases.
AcknowledgementsWe would like to thank the Dermatology, Radiology and Surgical department of Hospital Raja Perempuan Zainab II Kelantan for providing the skin biopsy histopathological specimen, CT scan of the abdomen and confirmatory tissue histopathological specimen respectively in the completion of this case report.
Ethical ConsiderationPatient consent form has been procured prior to the case report study.
Statement of AuthorshipAll authors have given approval to the final version submitted.
Author DisclosureAll the authors declared no conflicts of interest.
Funding SourceNone.
1. Sachithanandan S, Tzen CY, Raman K, Colao A. Neuroendocrine tumor symposium (GIST & NETs: A Multidisciplinary Journey). Oncology Tribune. Nov. 2010:2. http://www.mems.my/file_dir/20344153334d6c5f4d5d075.pdf.
2. Alkemade JA, van Tongeren JH, van Haelst UJ, et al. Delayed diagnosis of glucagonoma syndrome. Clin Exp Dermatol. 1999;24(6):455-7. http://dx.doi.org/10.1046/j.1365-2230.1999.00531.x. 3. Wermers RA, Fatourechi V, Wynne AG, Kvols LK, Lloyd RV. The glucagonoma syndrome: Clinical and pathologic features in 21 patients. Medicine (Baltimore).1996;75(2):53-63. http://dx.doi.org/10.1097/00005792-199603000-00002. 4. DeLellis RA , ed. Pathology and genetics of tumours of endocrine organs, 3rd ed. Vol. 8. IARC WHO Classification of tumours, 2004. 5. Butte JM, Montero PH, Solar A, Torres J, et al. Cervical metastases of glucagonoma in a patient with multiple endocrine neoplasia type 1: Report of a case. Surgery Today. 2008;38(12):1137-43. http://dx.doi.org/10.1007/s00595-008-3763-1. 6. Eldor R, Glaser B, Fraenkel M, Doviner V, et al. Glucagonoma and the glucagonoma syndrome-cumulative experience with an elusive endocrine tumour. Clin Endocrinol. 2011;74(5):593-98. http://dx.doi.org/10.1111/j.1365-2265.2011.03967.x. 7. Shi W, Liao W, Mei X, Xiao Q, et al. Necrolytic migratory erythema associated with glucagonoma syndrome. J Clin Oncol. 2010; 28(20):e329-e331. http://dx.doi.org/10.1200/JCO.2009.25.7113. 8. Stacpoole PW. The glucagonoma syndrome: Clinical features, diagnosis, and treatment. Endocr Rev. 1981; 2(3):347-61. http://dx.doi.org/ http://dx.doi.org/10.1210/edrv-2-3-347. 9. Koike N, Hatori T, Imaizumi T, Harada N, et al. Malignant glucagonoma of the pancreas diagnosed through anemia and diabetes mellitus. J Hepatobiliary Pancreat Surg. 2003:10(1):101-5. http://dx.doi.org/ 10.1007/s10534-002-0791-y. 10. Fenkci SM, Yaylali GF, Sermez Y, Akdam H, et al. Malign cystic glucagonoma presented with diabetic ketoacidosis: Case report with an update. Endocr Relat Cancer. 2005; 12(2):449-54. http://dx.doi.org/ 10.1677/erc.1.00957. 11. McGevna L, McFadden D, Ritvo J, et al. Glucagonoma-associated neuropsychiatric and affective symptoms: Diagnostic dilemmas raised by paraneoplastic phenomena. Psychosomatics. 2009;50(5):548-9. http://dx.doi.org/ 10.1176/appi.psy.50.5.548. 12. Mallinson CN, Bloom SR, Warin AP, Salmon PR, Cox B. A glucagonoma syndrome. The Lancet.1974; 304(7871): 1-5. http://dx.doi.org/10.1016/S0140-6736(74)91343-9. 13. van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: A clinical review. Eur J Endocrinol. 2004:151(5):531-7. http://dx.doi.org/10.1530/eje.0.1510531. 14. Remes-Troche JM, García-de-Acevedo B, Zuñiga-Varga J, et al. Necrolytic migratory erythema: A cutaneous clue to glucagonoma syndrome. J Eur Acad Dermatol Venereol. 2004;18(5):591-5. http://dx.doi.org/ 10.1111/j.1468-3083.2004.00981.x. 15. Teixeira RC, Nico MM, Ghideti AC. Necrolytic migratory erythema associated with glucagonoma: A report of 2 cases. Clinics. 2008:63(2):267-70. http://dx.doi.org/10.1590/S1807-59322008000200016. 16. Kitamura Y, Sato M, Hatamochi A, Yamazaki S, et al. Necrolytic migratory erythema without glucagonoma associated with hepatitis B. Eur J Dermatol. 2005;15(1):49-51. 17. Delaporte E, Catteau B, Piette F. Necrolytic migratory erythema-like eruption in zinc deficiency associated with alcoholic liver disease. Br J Dermatol.1997;137(6):1027-8. http://dx.doi.org/10.1111/j.1365-2133.1997.tb01583.x. 18. Castro PG, de León AM, Trancón JG, et al. Glucagonoma syndrome: A case report. J Med Case Rep .2011;5:402. http://dx.doi.org/10.1186/1752-1947-5-402. 19. O'Grady HL, Conlon KC. Pancreatic neuroendocrine tumours. Eur J Surg Oncol. 2008;34(3):324-32. http://dx.doi.org/10.1016/j.ejso.2007.07.209. 20. Kindmark H, Sundin A, Granberg D, Dunder K, Skogseid B, Janson ET. Endocrine pancreatic tumors with glucagon hypersecretion: A retrospective study of 23 cases during 20 years. Medical Oncology. 2007;24(3):330-7. http://dx.doi.org/10.1007/s12032-007-0011-2. 21. Taupenot L, Harper KL, O'Connor DT. The chromogranin-secretogranin family. New Engl J Med. 2003;348(25): 2579-80. http://dx.doi.org/10.1056/NEJM200306193482519. 22. Chastain MA. The glucagonoma syndrome: A review of its features and discussion of new perspectives. Am J Med Sci. 2001;321(5):306-20. http://dx.doi.org/ 10.1097/00000441-200105000-00003. 23. Norton J, Kivlen M, Li M, Schneider D, et al. Morbidity and mortality of aggressive resection in patients with advanced neuroendocrine tumors. Arch Surg. 2003;138(8):859-66. http://dx.doi.org/10.1001/archsurg.138.8.859. 24. Stabile BE. Islet cell tumors. The Gastroenterologist.1997;5(3):213-32. 25. Kheir SM, Omura EF, Grizzle W. Histologic variation in the skin lesions of the glucagonoma syndrome. Am J Surg Pathol. 1986;10(7):445-53. 26. Boden G. Insulinoma and glucagonoma. Semin Oncol. 1987;14(3):253-62. 27. El Darouty M, El Ela MA. Necrolytic migratory erythema without glucagonoma in patients with liver disease. J Am Acad Dermatol. 1996;34(6):1092. http://dx.doi.org/10.1016/S0190-9622(96)90299-6. 28. Chu QD, Al-kasspooles MF, Smith JL, et al. Is glucagonoma of the pancreas a curable disease? Int J Pancreatol. 2001;29(3):155-62. http://dx.doi.org/ 10.1385/IJGC:29:3:155. 29. Liu WJ, Zhao YP, Zhang TP, Liao Q, Cong L. Clinical experience in diagnosis and treatment of glucagonoma. Zhonghua wai ke za zhi. Chinese. 2009;47(5): 333-6. 30. Åkerström G, Hellman P, Hessman O, Osmak L. Surgical treatment of endocrine pancreatic tumours. Neuroendocrinology. 2004;80(Suppl. 1):62-6. http://dx.doi.org/10.1159/000080744. 31. Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-63. http://dx.doi.org/10.1200/JCO.2009.22.8510. 32. Alexander EK, Robinson M, Staniec M, Dluhy RG. Peripheral amino acid and fatty acid infusion for the treatment of necrolytic migratory erythema in the glucagonoma syndrome. Clin Endocrinol. 2002;57(6):827-31. http://dx.doi.org/10.1046/j.1365-2265.2002.01660.x. 33. Kulke MH, Benson AB, Bergsland E, Berlin JD, Blaszkowsky LS, Choti MA, et al. Neuroendocrine tumors. J Natl Compr Canc Netw. 201;10(6):724-64.Authors are required to accomplish, sign and submit scanned copies of the JAFES Author Form consisting of: (1) the Authorship Certification that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author, (2) the Author Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere, (3) the Statement of Copyright Transfer[accepted manuscripts become the permanent property of the JAFES and are licensed with an Attribution-Share Alike-Non-Commercial Creative Commons License. Articles may be shared and adapted for non-commercial purposes as long as they are properly cited], (4) the Statement of Disclosure that there are no financial or other relationships that might lead to a conflict of interest. For Original Articles involving human participants, authors are required to submit a scanned copy of the Ethics Review Approval of their research. For manuscripts reporting data from studies involving animals, authors are required to submit a scanned copy of the Institutional Animal Care and Use Committee approval. For Case Reports or Series, and Images in Endocrinology, consent forms, are required for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent. Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.