This case illustrates a patient with 3 diseases: diabetes mellitus type 1 (T1DM), alopecia universalis and common variable immunodeficiency disease (CVID). The occurrence of autoimmune disease in patients with common variable immunodeficiency has been well described in literature. Alopecia is not rare in patients with various autoimmune conditions. However, the presence of all 3 diseases in one individual has not been previously reported.
This is a 19-year-old Filipino male who presents with a 2-week history of polyphagia, polydypsia and polyuria and who was admitted for fever and chills with vomiting. There was body weakness, undocumented weight loss and blurring of vision.
After the age of 7 months, he was noted to have recurrent respiratory tract infections until the age of 3 years. At the age of 4 to 6 years old, he also experienced recurrent bouts of bronchitis and pneumonia occurring every 3 to 5 months for which he was treated with oral and occasional intravenous antibiotics. At age six, there was documentation of low immunoglobulin levels : IgA at 0.42 g/L (NV 0.9-4.45) and IgG at 1.0 g/L (NV 8-18); however, the IgM at 1.6 g/L (NV 0.7-2.8) was within normal limits. Gammaglobulin was administed at a dose of 5 ml intramusculary over each buttock for 7 doses. Infections became less frequent by the time he was 7 years old. However, at 10 years of age, there was note of patchy hair loss, later becoming generalized over a year; he was diagnosed with alopecia universalis at age 11. He continued to have frequent respiratory infections. He has a history of bronchial asthma and allergic rhinitis. He does not have diabetes nor hypertension. His last admission for pneumonia was 2 months prior to admission wherein persistent interstitial infiltrates were noted by his pulmonologist in a CT scan of the lungs. It was noted that he had haziness and reticulonodular densities in the anterior segment of the right upper lobe and anteromedial segment of the left lower lobe of the lungs despite full antibiotic coverage.
He is the younger of two siblings born to a healthy, non-consanguineous couple of Filipino descent. There is no family history of diabetes, autoimmune disease nor immunodeficiency. There is a family history of hypertension, endometrial cancer and stroke.
He does not smoke but is an occasional alcohol drinker.He denies illicit drug use. He denies sexual contact.
Physical examination revealed a coherent, fairly active, underweight male. He measured 173 cms in height and weighed 45 kg with a BMI of 15.1 kg/m 2
. He was slightly tachycardic at 104 bpm, normotensive and afebrile. He had generalized absence of the hair on the scalp, eyelashes, eyebrows, axilla, legs and arms, and sparse pubic hair. His skin was dry, especially on lower extremities. Except for pale palpebral conjunctivae and dry lips, his head and neck examination was normal. The thyroid gland was not enlarged and there were no palpable nodules. Likewise, his cardiac, lung and abdomen findings were normal. He had sparse pubic hair (Tanner stage 2) and his penile size was appropriate for age (Tanner 5). There was no edema nor cyanosis, and he had full and equal pulses. He was oriented to 3 spheres and his neurologic examination was normal.
Upon admission, his blood glucose was elevated at 588 mg/dl. Arterial blood gas showed uncompensated metabolic acidosis with a pH of 7.2 and a bicarbonate level of 8mEq/L. Urinalysis revealed glucosuria and ketonuria but was negative for protein. Serum ketones was at 6.6mmol/L. HbA1C was elevated at 8.5%. Further tests showed elevated anti-GAD65 at 59.48 kU/L(NV <10.00), islet cell antibody was normal at <5 JDF units (NV <5), and C-peptide level was lowat 0.26 ng/ml (NV 1.1-4.4).
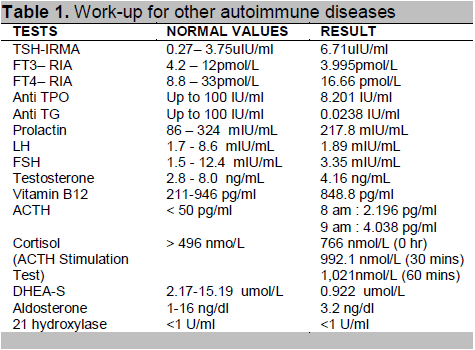
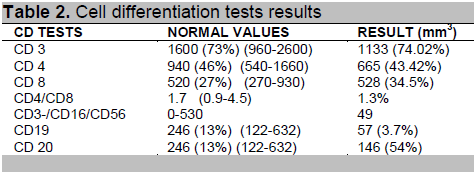
Further investigations revealed elevated TSH 6.71 uIU/ml (NV 0.27–3.75) and low FT3 3.99 pmol/L(NV 4.2–12), FT4, anti-TPO and anti-TG, prolactin, cortisol, LH, FSH and testosterone levels were normal (Table 1). Tests for immunodeficiency revealed low B cell counts, CD 19 is 57 mm3 (NV 246 mm 3) and CD 20 is 146 mm3 (NV 246 mm3). T cell (CD4 and CD 8) absolute counts were within normal range, the CD4/CD8 ratio is decreased at 1.3% (NV 1.7%;Table 2). Serum IgA and serum IgG were decreased at 0.19g/L (NV0.7-4.0 g/L) and 1.99g/L (NV 7-16.0 g/L) respectively while serum IgM level was normal.
Click here to download Table 1
Table 1. Work-up for other autoimmune diseases
Click here to download Table2
Table 2. Cell differentiation tests results
The discharge diagnosis was diabetic ketoacidosis secondary to diabetes mellitus type 1,Alopecia Universalis, rule out Polyglandular Autoimmune Syndrome and Primary Immunodeficieny Disease, Common Variable Immunodeficiency.
Clinical and laboratory findings were consistent with moderate diabetic ketoacidosis. He was given intravenous hydration and was started on insulin drip to control the hyperglycemia. The insulin drip was shifted to basal-bolus regimen when glucose levels were adequatedly controlled. Immunoglobulin (IVIg) replacement therapy was initiated one month after the patient was discharged.
The patient was discharged with good glucose control using basal-bolus insulin regimen. Monthly intravenous immunoglobulin infusions was recommended and he was advised regarding good nutrition and hygiene to avoid infections. Further molecular studies to determine the exact genotype of CVID was also advised. Regular monitoring for emergence of other types of autoimmune conditions will be done on outpatient basis. Regular monitoring of endocrine hormones was also recommended because of the polyglandular autoimmune disease.
The diagnosis of immunodeficiency must be considered in a patient with recurrent pneumonia and concomitant autoimmune disease. This patient experienced recurrent pneumonia and upper respiratory tract infections after the age of 7 months when protective maternal antibody levels have begun to wane. According to the Jeffrey Modell Foundation screening tool: The 12 Warning Signs of Immunodeficiency,1 the presence of 2 bouts of pneumonia and 8 or more bouts of upper respiratory tract infections in a year warrants investigation for immunodeficiency. The patient presented with clinical features of immunodeficiency and low quantitative immunoglobulin levels of serum IgG and IgA.B cell enumeration via flow cytometry revealed low pre-B cell count (CD19) and low mature B cell counts (CD20) which indicate a humoral type of immunodeficiency. Although T cells counts (CD4 and CD8) were within normal values, there was a slightly decreased CD4/CD8 ratio suggesting a relative deficiency of CD 4 cells in spite of normal absolute counts. These findings, in the background of concomitant autoimmune diseases (diabetes mellitus and alopecia universalis) clinched the diagnosis of Common Variable Immunodeficiency (CVID), the most commonly encountered symptomatic primary immunodeficiency. CVID is a clinically heterogenous disorder presenting at any age, characterized by recurrent bacterial infections, hypogammaglobulinemia, impaired antibody responses despite the presence of B cells and normal or near-normal T cell-immunity.1 CVID is caused by defective antibody production with low levels of serum immunoglobulin which leads to increased susceptibility to infection.2 An expert opinion from our geneticist3 revealed that the factors that contribute to increased autoimmunity in patients with CVID include genetic predisposition, innate and adaptive immunity deficiencies, variable degrees of immune dysregulation and possible failure in central and peripheral mechanisms of tolerance induction or maintenance which can all lead to persistent or recurrent infections. She further elaborated that the important issues in the genetics of CVID are the following: this condition has an estimated prevalence of ∼1 case per 30,000 among Caucasians; and no Asian prevalence rates have yet been established but there are a number of unreported cases; onset of symptoms typically appears after puberty, and the diagnosis is usually made in the second or third decade of life; there is no clear pattern of inheritance, majority of the cases are sporadic; although, familial occurrence has been reported in 20- 25% of cases and a positive family history for humoral immunodeficiency is evident in 10 to 20%. When more than one family member is affected with CVID, approximately 5% of the patients have a concurrent IgA deficiency. Most of the familial cases show an autosomal dominant mode of inheritance; members of the family who are affected may either have CVID, selective IgA deficiency or intermediate states of humoral immunodeficiency.4 A three generation family history was elicited on this patient during his family’s visit at the geneticist’s clinic, however, no clear inheritance pattern was observed She also shared that to determine the molecular genetics of heritable CVID, there are three genetic tests we can offer this patient namely: TACI-associated CVID, ICOS-deficiency and CD-19-deficiency. Knowing the genetic defect may be useful because it can influence the type of immune dysregulation observed and the underlying genetic defect will likely be important in determining long term outcomes. 3
The pathogenesis of CVID is yet unknown; it is thought to be due to several molecular defects which are polygenic in nature.5 B cells are unable to differentiate into plasma cells and there may also be T cell and monocyte/macrophage defects.2 Patients with CVID may suffer from several sequelae of immune dysregulation. They are prone to recurrent sinopulmonary infections leading to bronchiectasis. Inflammatory bowel diseases and nodular lymphoid hyperplasia of the small bowel are common complications. The development of non-caseating granulomas and malignancy (lymphomas) may occur with high incidence.5 However, the presence of autoimmune disease together with immunodeficiency is typical of CVID. Around 20-25% of patients with CVID develop autoimmune disease.1 The interrelations between genetic defects, immunological abnormalities and clinical phenotypes will improve our understanding of CVID and its pathogenesis.
The patient’s immunodeficiencywas also associated with alopecia universalis. Alopecia universalis involves rapid loss of all hair including eyebrows and eyelashes. When the body's immune system attacks hair follicles, it causes patchy, widespread or total hair loss. This may be caused by a gene called ULBP3, which belongs to a class of proteins known as natural killer (NK) cell ligands poised to injure the hair follicle.6
Type 1 Diabetes Mellitus, an autoimmune disease, results from selective destruction of the β cells in the pancreatic islets.7 Βeta cells are destroyed by an aggressive autoimmune process mediated by infiltration of CD4+ and CD8+ T cells, as well as macrophages, resulting in insulitis. 8 The patient has low C-peptide and an elevated anti-GAD65, the most sensitive antibody for the detection of type 1 diabetes.9 Autoimmune diseases are commonly the first manifestation of immune deficiency.2 Approximately 20-30% of CVID patients show autoimmune phenomena and/or develop manifest autoimmune disease.10 However, the pathogenesis of autoimmunity in CVID remains obscure. How autoantibodies are produced against specific tissues in a state of impaired antibody production is unclear.2 The dysregulation of the immune system in CVID often seems paradoxical, while antibody production in response to pathogens and vaccines is severely impaired or even lacking, the generation of autoantibodies may, at the same time, be in excess.10
The presence of two autoimmune diseases in the same patient prompted further investigation for polyglandular autoimmune syndrome. According to Van den Driessche,11 up to one third of patients with diabetes mellitus type 1 develop an autoimmune polyglandular syndrome. Autoimmune thyroid disease (Hashimoto’s or Graves’ disease) is seen in 15 to 30%; 5 to 10% are diagnosed with autoimmune gastritis and/or pernicious anemia; 4 to 9% present with celiac disease (CD); 0.5% have Addison’s disease and 2 to 10% have vitiligo.11 The patient’s TSH is elevated at 6.71uIU/ml with normal FT4, interpreted as subclinical hypothyroidism probably due to autoimmune thyroiditis. Further tests showed normal anti-TPO and anti-thyroglobulin. Thyroid function test repeated a month after admission was normal. Autoimmune mediated thyroid disease was ruled out at this point due to normal antibody tests. A transient thyroiditis of infectious origin could have caused the TSH elevation.
Pernicious anemia was unlikely since the patient’s vitamin B12 level was normal and he did not have anemia. Neither were there symptoms of celiac disease. Addison’s disease was also ruled out since baseline cortisol levels, ACTH, ACTH stimulation test and 21-hydroxylase were all normal. Clinically, the patient did not present with signs of adrenal insufficiency. Vitiligo is also an autoimmune-mediated hypomelanotic disorder that was not observed in our patient.
Polyglandular Autoimmune Syndrome (PAS) represents different clusters of autoimmune disorders. These rare endocrinopathies are characterized by the coexistence of at least two glandular autoimmune mediated diseases.12 In this case, PAS I and II were ruled out due to absence of adrenal insufficiency, PAS III was also ruled out due to absence of an autoimmune thyroid disease. Neufeld has proposed another category,13 PAS IV, which usually involves two or more autoimmune endocrine diseases and does not follow the typical pattern for the other types of PAS. PAS IV is not as common as the other three PAS disorders and is diagnosed based on which endocrine glands are not affected, rather than by which ones are.13 This patient presenting with T1DM and alopecia universalis is likely to have polyglandular autoimmune syndrome type IV.
A link between autoimmunity and immunodeficiency has long been established. It seems that in this patient, immunodeficiency was present early in life, documented by recurrent respiratory infections and low antibody levels. However, the coexisting generalized alopecia and type 1 diabetes prove that in addition to immunodeficiency, this patient may also have an autoimmune disease process. The management of this patient is focused on replacement of immunoglobulin through monthly intravenous immunoglobulin infusions at 400 mg/kg/dose.The main goal is to have a significant reduction of life-threatening bacterial infections and prevention of end-organ damage (lungs, GIT) caused by the infections and immune dysregulation. IVIG treatment does not only provide antibodies passively but induces proliferation and antibody production in B cells of CVID patients. Due to its immunomodulating effects, it has an active role in regulating autoimmune and inflammatory responses of T and B cells.14 The desired immunoglobulin G level is between 5 and 7 gm/L. After the fourth immunoglobulin infusion, the patient already maintained a through level of 7.1 gm/L and there were no further hospital admissions for severe infections. The prognosis of patients with CVID is improved with the use of immunoglobulins and antibiotics. The overall mortality in 7 years is very low, if good control of the disease is maintained. The coexisting autoimmune disorder, necessitates regular monitoring of antibody levels and continued surveillance for the development of other autoimmune diseases and malignancy is warranted.
From the foregoing clinical case presentation and discussion, the auhtors outline several learning points:
· Common variable immunodeficiency (CVID) is the most common symptomatic primary immunodeficiency.
· The spectrum of immunological abnormalities in CVID affects T cells, B cells and antigen-presenting cells.
· CVID is characterized by hypogammaglobulinemia and recurrent bacterial infections.
· CVID may be complicated by autoimmunity such as diabetes mellitus type 1 and alopecia.
· Lifelong monthly intravenous immunoglobulin replacement therapy is the mainstay of CVID therapy.
· Regular follow-up and adjunctive therapy help to control the complications and sequelae of immundeficiency and autoimmunity is warranted.
References
1. Steihm ER, Ochs HD et al eds. Immuologicdisordersin infants and children, 5th edition Philadelphia PA; Elsevier Saunders 2004.
2. Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency, Curr Allergy Asthma Rep. September 2009 ; 9(5):347–352.
3. Cutiongco-Dela Paz, E. 66th Inter-hospital Grand rounds of the Philippine Society of Endocrinology and Metabolism, Makati Medical Center , May 2011.
4. Vorechovsky I, Zetterquist H, Paganelli R et al. Family and linkage study of selective IgA deficiency and common variable immunodeficiency. Clin. Immunol. Immunopathol. 1995;77:185-192.
5. Chapel H and Cuninnham-Rundles, C. Br J Haematol 2009;145(6):709-727.
6. Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. Jul 1 2010;466(7302):113-117.
7. American Diabetes Association: Standards of Medical Care in Diabetes—2011, Diabetes Care. January 2011; 34(1): S11-S61.
8. Jameson JL, De Groot LJ. Endocrinology, Adult and Pediatric 6th ed. 2010.
9. Gardner DG, Shoback D. Greenspan’s Basic and Clinical Endocrinology, 9th ed. McGraw Hill Companies, Inc. 2011:588.
10. Bergbreiter A, Salzer U. Common variable immunodeficiency: A multifaceted and puzzling disorder. Expert Rev ClinImmunol. 2009;5(2):167-180.
11. Van den Driessche A, Eenkhoorn V, Van Gaal L, De Block C. Type 1 diabetes and autoimmune polyglandular syndrome: A clinical review. The Netherlands Journal of Medicine, December 2009; 67 (11) 376-387.
12. Kahaly, GJ. Polyglandular autoimmune syndromes. European Journal of Endocrinology.2009;161: 11–20.
13. Neufeld M, Maclaren N, Blizzard R. Polyglandular autoimmune diseases.Pediatr Ann. 1980;9(4):154-62.
14. Bayry J et al. J Autoimmun. Feb 2012; 36 (1):9-15.
Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.