A Rare Large Cell Neuroendocrine Carcinoma in a 72-year-old Man
Teodora Amor N. Evora and Michael L. Villa
Teodora Amor N. Evora, M.D.
Diabetes, Thyroid and Endocrine Center
12th fl., Cathedral Heights Building,
St. Luke's Medical Center, E. Rodriquez Ave.,
Quezon City, Philippines
Tel. No.: 7230101 (local 5210)
Fax No.: 7230101 (loval 5210)
Mobile No.: 0922-8372185
e-ISSN 2308-118x
Printed in the Philippines
Copyright © 2011 by the JAFES
Received September 16, 2010. Accepted February 10, 2011.
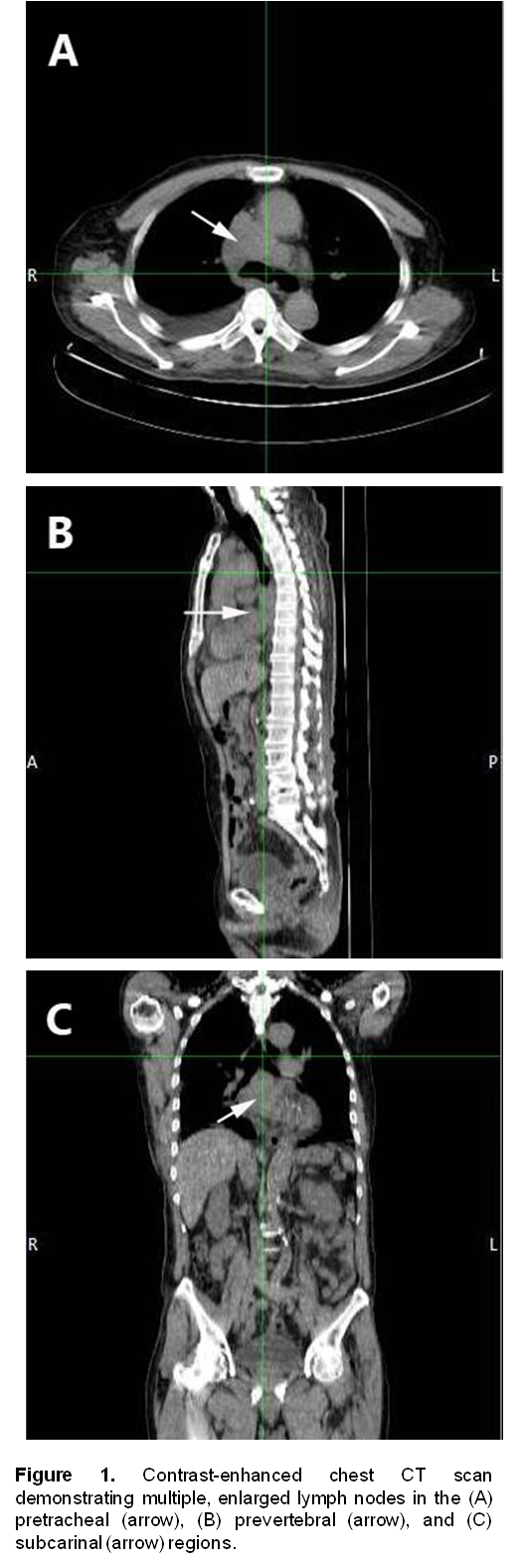
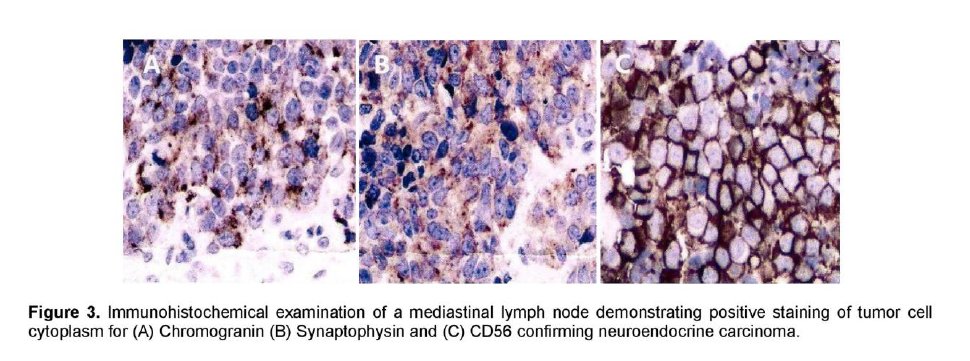
A 72-year-old male, previous smoker presented with cough and shortness of breath of one week duration. His chest x-ray showed linear densities in the right paracardiac and both lung bases. Contrast-enhanced CT scan uncovered multiple, enlarged soft tissue masses in the pretracheal, right paratracheal, subcarinal and right paravertebral regions (Figure 1). There were also reticular ground glass opacities scattered in both lung bases and periphery. No endobronchial mass was seen. The thyroid, adrenal glands, pancreas, liver and the rest of the visceral organs were normal. Mediastinoscopy with excision biopsy was done. Microscopic examination revealed large tumor cells arranged in solid sheets or nests (Figure 2). The differential diagnosis included diffuse large cell lymphoma and poorly-differentiated carcinoma believed to be primary lung versus mediastinal cancer. Positive immunohistochemical staining of tumor cell cytoplasm for Chromogranin, Synaptophysin, and CD 56 (Figure 3), and negative for cytokeratins confirmed the diagnosis of Large Cell Neuroendocrine carcinoma. Whole body [18F]-Fluorodeoxyglucose (FDG) PET-CT scan was done to locate the primary tumor and delineate the extent of disease.
Click here to download Figure 1
Figure 1. Contrast-enhanced chest CT scan demonstrating multiple, enlarged lymph nodes in the (A) pretracheal (arrow), (B) prevertebral (arrow), and (C) subcarinal (arrow) regions.
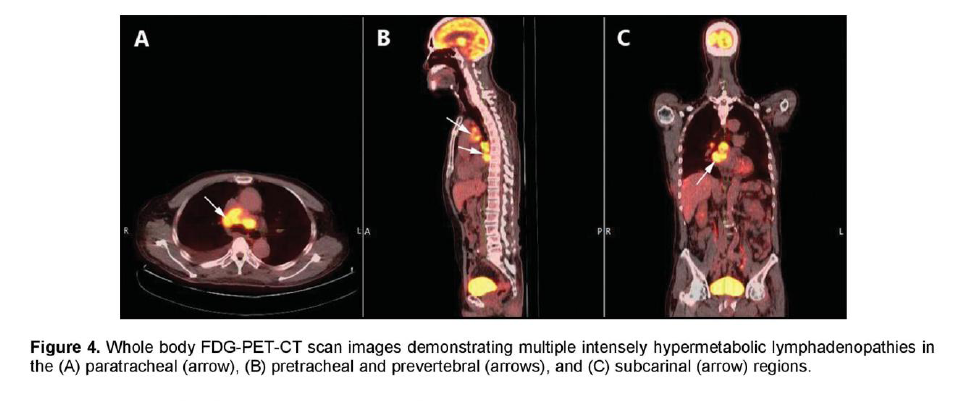
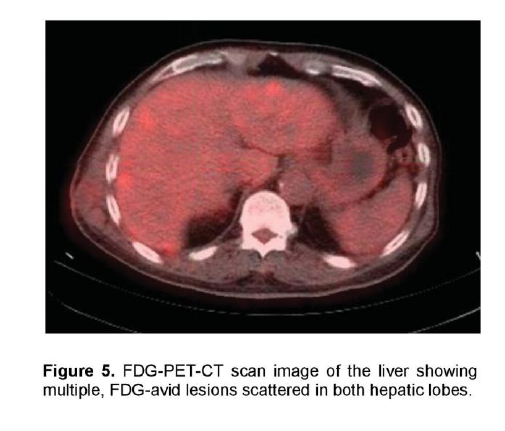
There was a mildly enhancing 1.3 x 1.3 cm mass in the right temporal lobe with FDG uptake similar to that of white matter. The previously seen mediastinal and right hilar lymphadenopathies had intense FDG uptake with standardized uptake value (SUV) up to 7.3 g/ml (Figure 4). In contrast, the previously noted reticular ground glass opacities in both lungs had low-grade uptake on PET. There were multiple hypermetabolic lesions in both hepatic lobes with SUV up to 5.4 g/ml (Figure 5) which appeared normal on the initial CT scan. A 24-hour urinary 5-hydroxyindoleacetic acid (5HIAA) was four times elevated, indicative of tumor secretory activity. He underwent chemotherapy with Carboplatin, Etoposide and Topotecan and radiation therapy with complete disappearance of the brain lesion but with further derangement in liver function.
Click here to download Figure 2
Figure 2. Photomicrographs of a mediastinal lymph node exhibiting (A) proliferation of tumor cells arranged in a diffuse nest pattern (H&E, 40x magnification); and (B) tumor cells with enlarged hyperchromatic and pleomorphic nuclei (arrows) and scanty cytoplasm (H&E, 400x magnification).
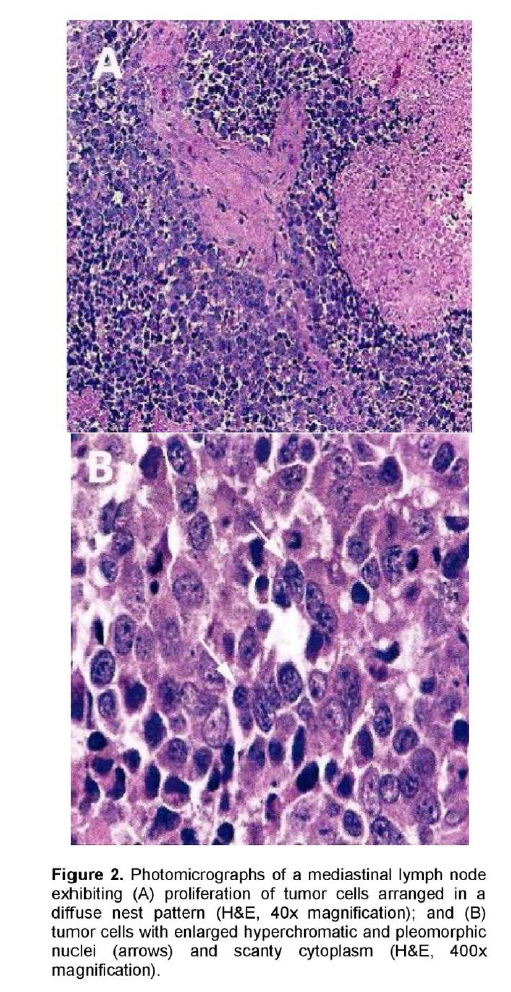
Figure 2. Photomicrographs of a mediastinal lymph node exhibiting (A) proliferation of tumor cells arranged in a diffuse nest pattern (H&E, 40x magnification); and (B) tumor cells with enlarged hyperchromatic and pleomorphic nuclei (arrows) and scanty cytoplasm (H&E, 400x magnification).
Neuroendocrine tumors (NET) are rare, solid malignant tumors that arise from dispersed neuroendocrine cells found throughout the body. According to the 2004 analysis of Surveillance, Epidemiology and End Results (SEER) database, the age-adjusted incidence of NET is of 5.25 cases per 100,000 people. 1 The majority of NETs arise within the gastroenteropancreatic system while the bronchial tract represents the second most frequent primary site.2 NETs with an undetected primary tumor have been noted in more than 4% of cases in several studies.2 A primary tumor site could not be found in 4,753 (13%) out of 35,618 NET in the SEER database analysis.1 NETs may produce a variety of clinical syndromes depending on a multitude of peptides and hormones being secreted. However, most NETs are clinically silent or elicit non-specific symptoms such that diagnosis is often delayed and with metastatic disease at presentation.1
The cornerstone of diagnosis of NET is histopathology including immunohistochemistry.2 Immunohistochemistry is most important for verification of the neuroendocrine nature of the tumor. It is also crucial in characterizing the proliferative potential of the tumor because this has been shown to be of prognostic value. Chromogranin A and/or Synaptophysin positivity are considered sufficient for the diagnosis.3 A 24 hr urine 5HIAA is a good biomarker of tumor secretory activity and may be predictive of reduced survival.2
Several imaging techniques are available for localization of tumor and monitoring disease progression. Somatostatin Receptor Scintigraphy (SRS) has a high sensitivity of 86-95% and should be the initial imaging procedure to localize and establish the stage of disease.4 However, the difficulty and expense of obtaining an SRS is prohibitive. FDG-PET scanning may be useful in finding a primary tumor and identifying poorly-differentiated anaplastic tumor.4
Treatment of metastatic neuroendocrine carcinoma of unknown primary site depends on the pathologic categorization as to whether the tumor is well-to moderately differentiated (large cell tumors) or poorly differentiated (high grade, anaplastic, small cell tumors).5 SRS is carried out to indicate the presence of somatostatin receptors and imply likelihood to respond to a long-acting somatostatin analogue. If the SRS is negative, other treatment options include hepatic artery chemoembolization (HACE), radioactive microspheres (SIRS) therapy, or conventional chemotherapy considering some of the newer agents, such as tyrosine kinase inhibitors.3
Our patient presented with a rare Large Cell Neuroendocrine carcinoma that has metastasized to the mediastinal and hilar lymph nodes, liver and brain but whose primary tumor was undetected. A possible primary lung cancer was highly considered. There was tumor progression 2 months after chemotherapy and radiation therapy. Our case may benefit from a SRS for further tumor localization and likelihood to respond to somatostatin analogue.
Click here to download Figure 3
Figure 3. Immunohistochemical examination of a mediastinal lymph node demonstrating positive staining of tumor cell cytoplasm for (A) Chromogranin (B) Synaptophysin and (C) CD56 confirming neuroendocrine carcinoma.
Click here to download Figure 4
Figure 4. Whole body FDG-PET-CT scan images demonstrating multiple intensely hypermetabolic lymphadenopathies in the (A) paratracheal (arrow), (B) pretracheal and prevertebral (arrows), and (C) subcarinal (arrow) regions.
Click here to download Figure 5
Figure 5. FDG-PET-CT scan image of the liver showing multiple, FDG-avid lesions scattered in both hepatic lobes.
1. Yao J, Hassa M, Phan A, et al. One hundred years after “Carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72.
2. Modlin I, Lye D, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-59.
3. Mamikunian G, Vinik A, O’Dorisio T, Woltering E, Go V. Neuroendocrine Tumors, A Comprehensive Guide to Diagnosis and Management. 2009; Inter Science Institute: 4th ed.
4. Balon H, Goldsmith S, Siegel B, et al. Society of Nuclear Medicine Procedure Guideline for Somastatin Receptor Scintigraphy with In-111 Pentetreotide. J Nucl Med. 2001;42:1134-8.
5. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Neuroendocrine Tumors. 2010. www.nccn.org. Accessed Feb 14, 2011.
Authors are required to accomplish, sign and submit scanned copies of the JAFES Declaration that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere.
Consent forms, as appropriate, have been secured for the publication of information about patients; otherwise, authors declared that all means have been exhausted for securing such consent.
The authors have signed disclosures that there are no financial or other relationships that might lead to a conflict of interest. All authors are required to submit Authorship Certifications that the manuscript has been read and approved by all authors, and that the requirements for authorship have been met by each author.