Hypophosphatemic osteomalacia comprises hereditary and acquired disturbances. Main syndromes described as causing this disease are x-linked hypophosphatemic rickets (XHR), Autosomal Dominant Hypophosphatemic Rickets (ADHR) and Fibrous dysplasia.
TIO is an acquired condition and is related to mesenchymal or mixed connective tissue tumour(s) that markedly expresses FGF23 RNA protein, which cannot be adequately degraded by specific endopeptidases. Other clinical syndromes have been also associated with FGF23 overproduction, such as NF–1, Mc Cune–Albright Syndrome (MAS).[1]
The FGF23 protein inhibits type II a and II c sodium – phosphate co-transport in proximal convoluted tubules and inhibits renal 1-α hydroxylation of vitamin D.
Although these conditions exhibit different etiopathogenic mechanisms, they share the same pathophysiological pathways i.e., secretion of FGF23 that acts on proximal tubules, and increases renal excretion of phosphate, leading to hyperphosphaturia, and hypophosphatemia with impaired vitamin D metabolism, as FGF23 inhibits 1-alpha hydroxylase.[2][3]
The diagnosis is often delayed, as the patient presents with multiple complaints, such as bone and muscle pain and muscle weakness. The tumour itself is small and slow growing, and often missed during the clinical examination. As clinical and biochemical changes of TIO are potentially reversible by tumour removal, its localization is essential.
We report a case of chronic hypophosphatemic osteomalacia, characterized by severe muscle weakness, pain and multiple bone fractures. Our aim is to register the case, and also discuss the diagnostic approach and therapeutic management of TIO.
CASEA 57-year-old Indian woman, presented with the chief complaints of bilateral leg pains on walking since 15 months, limping to either side since 3 months and currently walking with support of a stick since 1 month. The patient also complained of pain in both upper limbs since 1 month. She had past history of Chikungunya infection 4 years ago and history of left superficial parotidectomy in 2011. Before coming to our OPD, she was treated for Rheumatoid Arthritis, evaluated for multiple myeloma and connective tissue disorder due to intense pain in lower limbs. She had received oral calcitriol before coming to hospital. However, the patient was not given phosphorous supplement. There was no family history of metabolic bone disease. There was no history of loss of weight, decreased appetite, and swelling or mass anywhere in the body except a left parotid enlargement.
On physical examination, the patient had antalgic gait, and left parotid enlargement 3 x 4 cm. She had localized tenderness over multiple ribs and multiple tender points over the lower limbs. She had left sided lower motor neuron facial palsy. Motor strength in the lower limbs was grade 5, but she had intense pain (numerical response scale 8/10) in both the feet and right hip.
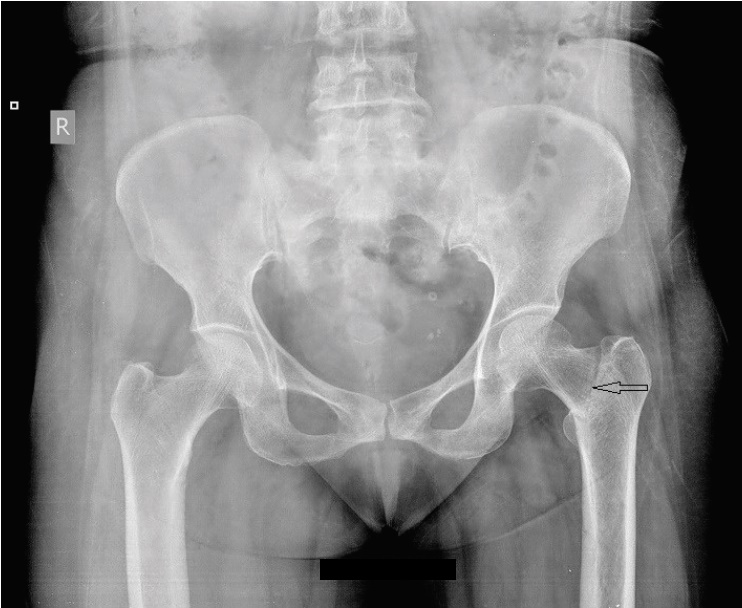
Initial laboratory testing revealed a calcium 9.6 mg/dl (8.8–10.2 mg/dl), phosphorus level 2 mg/dl, alkaline phosphatase was marginally elevated 179 U/L, creatinine 0.6 mg/dl, SGOT 22.1(U/L) SGPT 23.8(U/L) PTH 5.6 pg/ml, and vitamin D 17.5 ng/ml. Complete blood count, liver function tests, renal function tests, TSH, serum protein electrophoresis and CPK were normal (Table 1). We were unable to measure 1,25 vitamin D as the test was not available. The patient’s ANA and Rheumatoid factor was negative. Her EMG and Nerve conduction studies were normal.
Click here to download Table 1Table 1.Laboratory test results
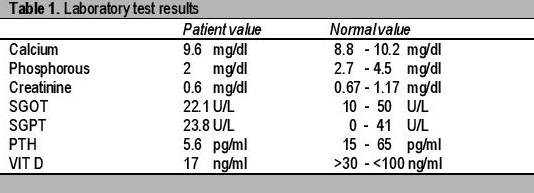
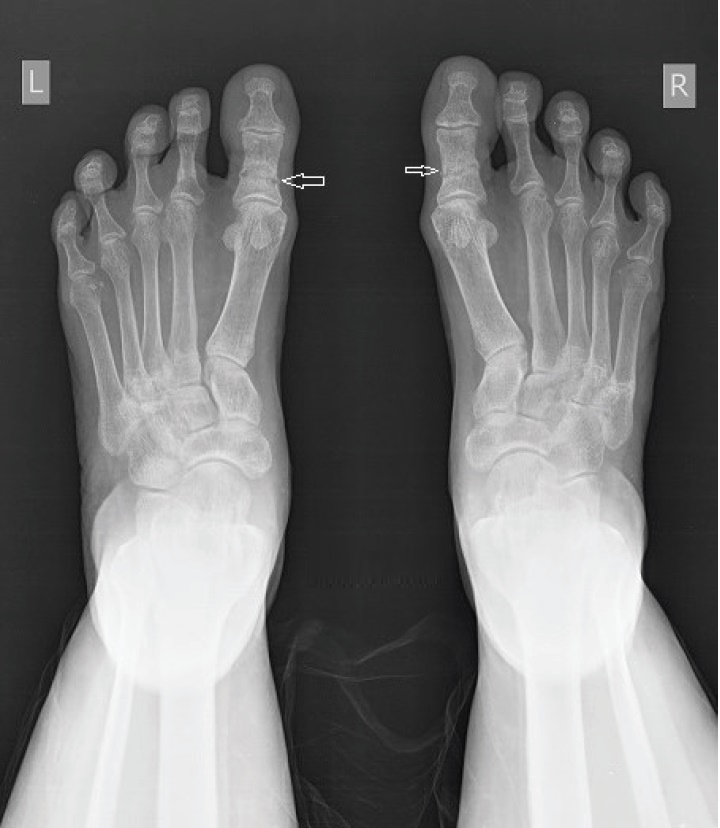
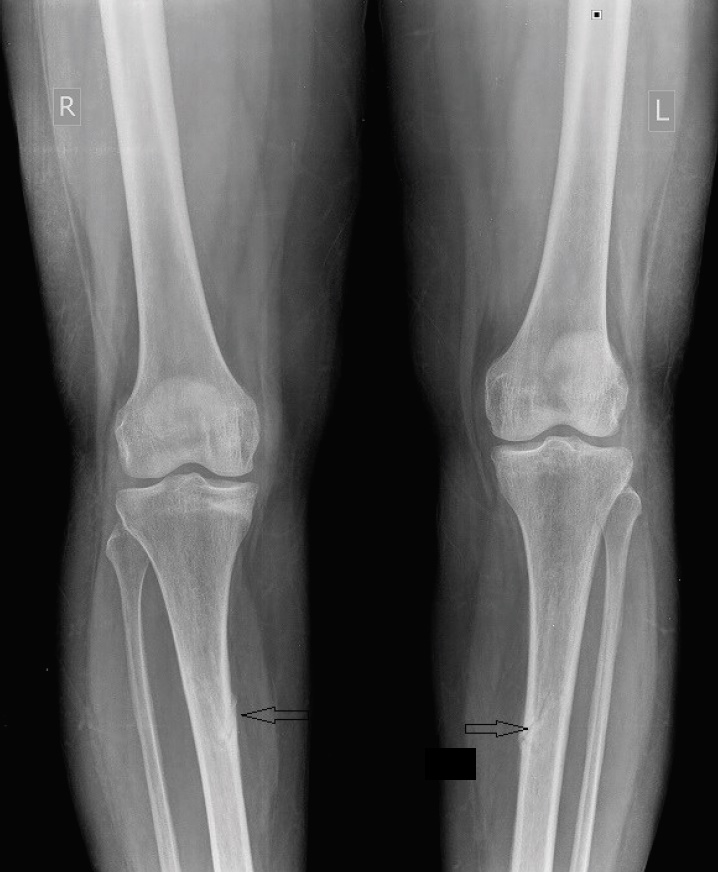
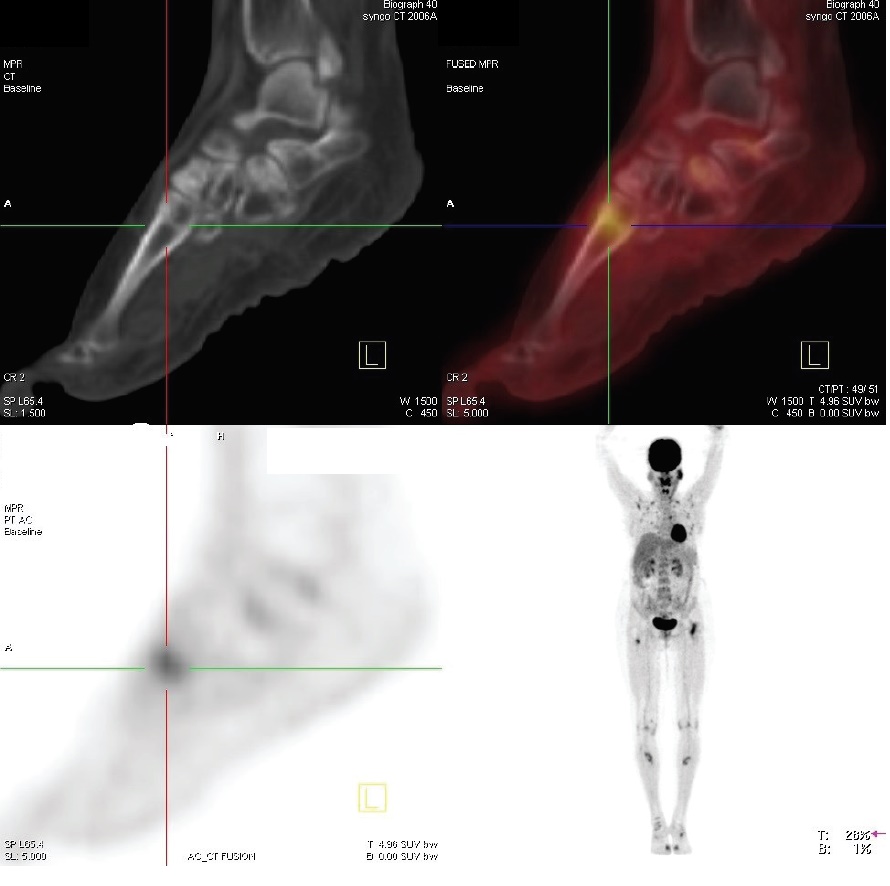
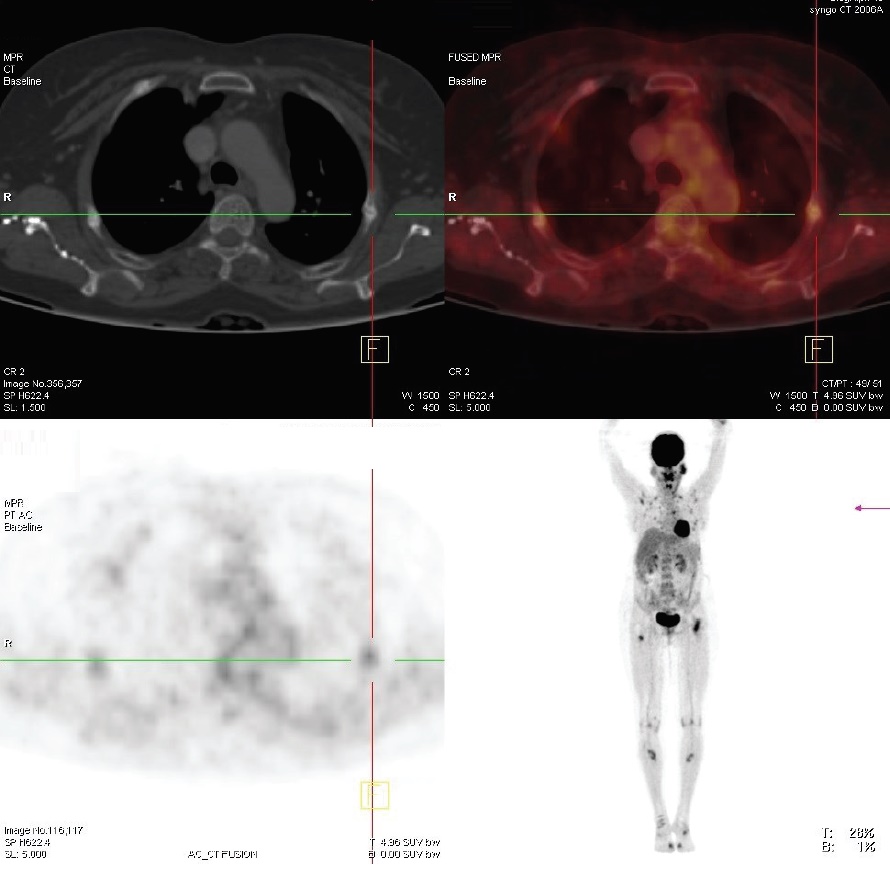
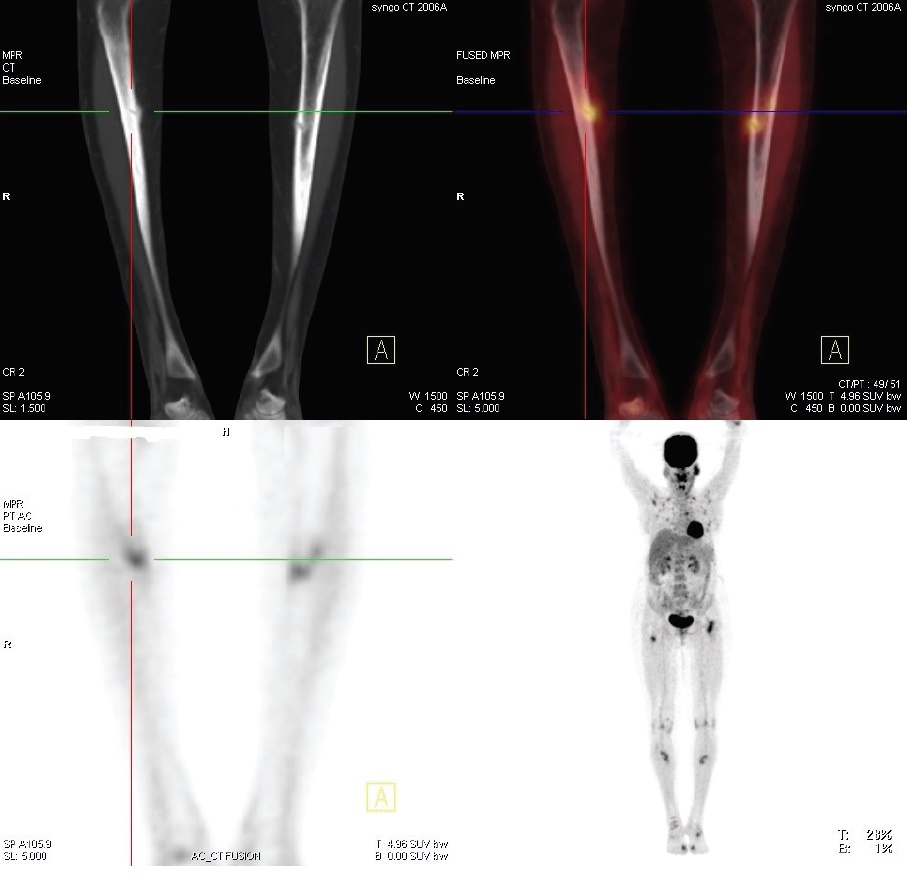
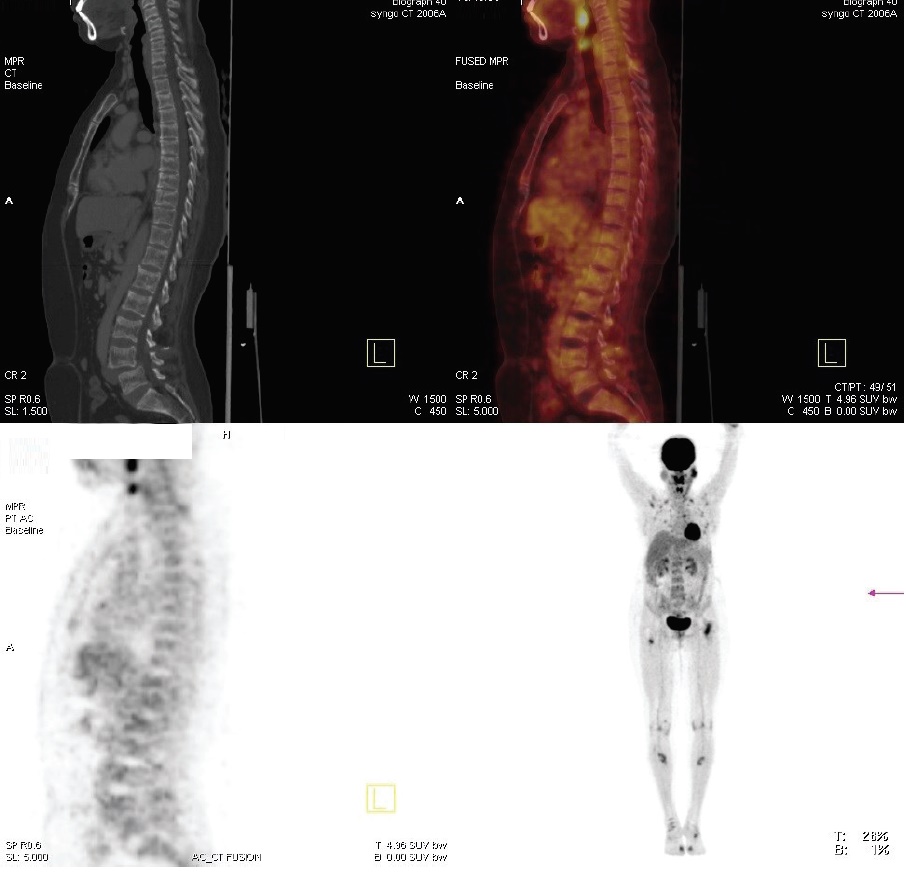
Radiological studies showed bilateral tibial shaft fractures, multiple fractures in bilateral tarsals (Figures 1 to 3). A PET Scan showed multiple insufficiency fractures of bilateral tibia, neck of right femur, metacarpal bones and scapula (Figures 4 to 7).
Click here to download Figure 1Figure 1.X-ray showing metatarsal fracture.
Click here to download Figure 2
Figure 2.X-ray showing bilateral midshaft tibia fracture.
Click here to download Figure 3
Figure 3.X-ray showing neck femure fracture on left side.
Click here to download Figure 4
Figure 4.PET scan showing uptake over fracture part.
Click here to download Figure 5
Figure 5.PET scan showing fracture of ribs.
Click here to download Figure 6
Figure 6.PET scan showing fracture of tibial shaft.
Click here to download Figure 7
Figure 7.PET scan showing multiple fractures.
Instead of 24-hour urinary phosphorous, levels of phosphorus and creatinine were measured from a spot urine sample and tubular re-absorption of phosphorus (TRP) was calculated. Walton and Bijovet Nomogram was then used to estimate the TMP/GFR, which was 1.6, suggestive of renal phosphate wasting. The patient’s FGF23 was 500 RU/ml.
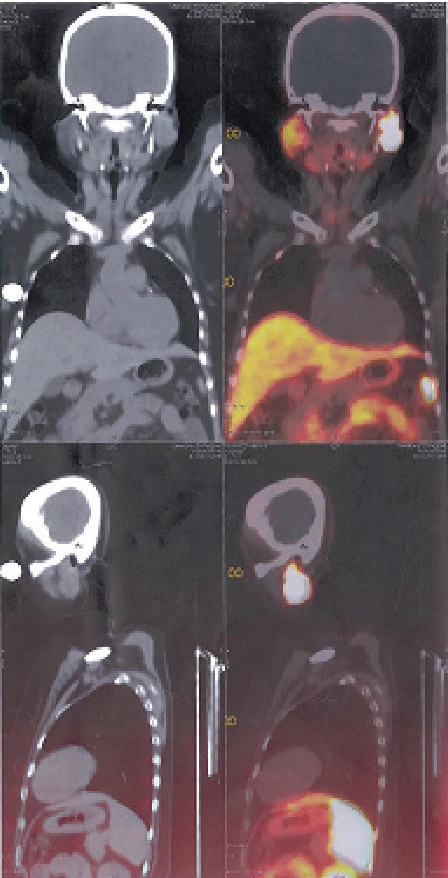
Since the parotid tumour also has a mesenchymal component, it was thought to be a source producing FGF23. (68)Ga-DOTATE scan was done which showed increased uptake over left parotid, SUV (Standard uptake values) max – 97.5, other areas, had normal physiological uptake (Figure 8).
Click here to download Figure 8Figure 8.(68)Ga-DOTATE scan showing coronal and sagittal view of mass in left parotid.
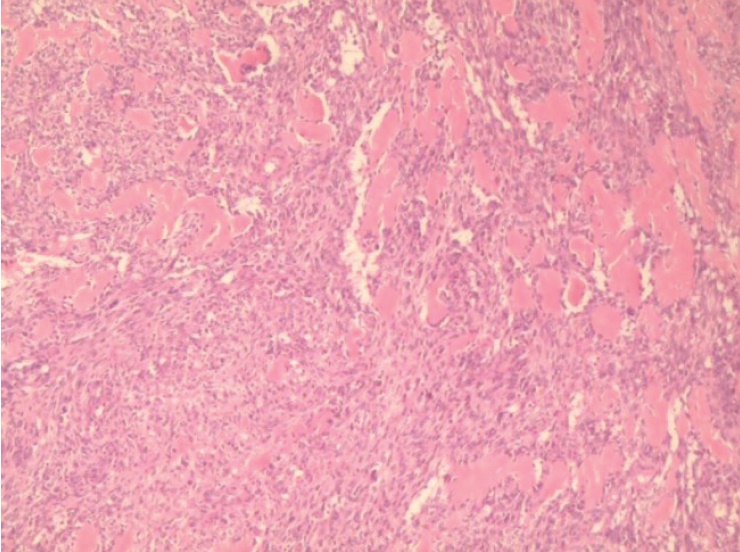
The patient was started on oral phosphate supplement 2-3 gms/ day, and her symptoms improved from baseline, but serum phosphorous remained on lower side. The patient underwent left parotid resection. The excised left parotid gland measured 4 x 3 x 2.5 cm and on cut section there was a grayish white ill-defined firm and focally hard solid tumour measuring 2.8 x 2.2 x 2 cm. A similar nodule measuring 0.5 cm in diameter was found in the adjacent parotid parenchyma. The rest of the parotid was unremarkable. The histopathology showed cellular myoepithelial rich pleomorphic adenoma of the parotid gland. Focal nucleal pleomorphism was noted. Mitotic count was low (Figures 9 and 10). Immuno-staining of the tumour tissue for FGF23 could not be done as this test was not available.
Click here to download Figure 9Figure 9.Cellular spindle cell tumour with deposition of basement membrane like eosinophilic material (20x).
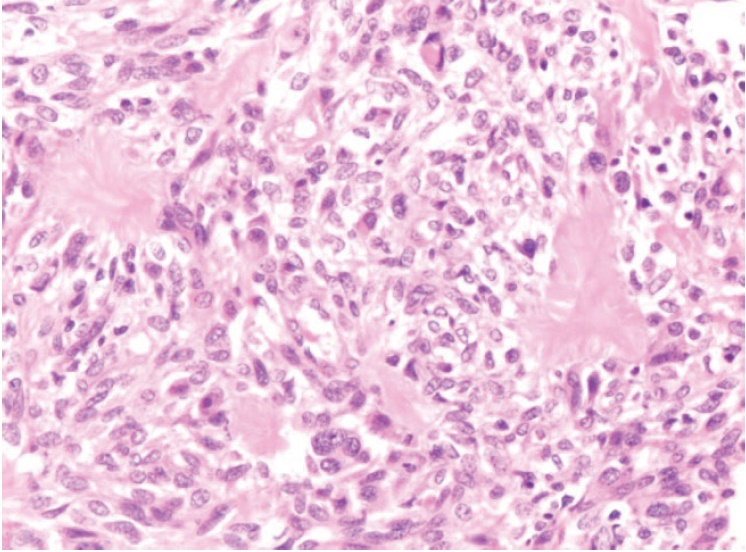
Click here to download Figure 10
Figure 10.High power view of the tumour showing increased cellularity and mild nuclear pleomorphism. However, the mitotic activity is low (40x).
The patient’s symptoms improved after resection, and serum phosphorous done 1 week after resection showed a normal phosphorus.
Tumour induced osteomalacia (TIO) is an acquired disorder of renal phosphorus wasting. It is associated with tumour producing phosphaturic factor leading to phosphaturia, low serum phosphorus levels and low or inappropriately normal serum calcitriol levels. Hypophosphatemia is normally a stimulus for production of calcitriol, and the level of normal vitamin D is abnormal in the context of low serum phosphorus level.[4][5][6]
Clinical symptoms include the gradual onset of muscle weakness, fatigue and bone pain especially from ankle, legs, hips and back.3-5 Insufficiency fractures and proximal muscle weakness causes pain and inability to walk; so the patient becomes slowly bed bound. Our patient had multiple fractures of tibia, femur and metatarsals due to which she had gradually become bed bound. She was diagnosed and treated as having Rheumatoid Arthritis due to the intense pain in her legs while walking.
The phosphaturic peptides are known as phosphatonins and FGF23 is the most extensively described. FGF23 has an important role in maintaining phosphorus homeostasis.[3]-[6] In TIO, the level of FGF23 is high and results in phosphate wasting through the urine.[7] If the responsible tumour is surgically removed, the abnormalities of phosphorus wasting and vitamin D metabolism are rapidly corrected. According to White, PTH which decreases renal phosphorus reabsorption is usually within normal range in TIO patients.[8] Hence in our patient, due to increased FGF23, the serum PTH must have been low. Other studies have supported this hypothesis by showing that implantation of tumour tissue into nude mice resulted in increased urinary phosphorus excretion.[9]
When TIO tumours and several control tissue were tested by northern blot for the presence of FGF23 transcripts, it was determined that FGF23 was present in a tumour lysate and it was confirmed by western blot analysis, with an anti human FGF antibody. The mice receiving implanted transcripts from tumour also showed growth retardation, kyphosis, osteomalacia and marked decrease in renal 1α hydroxylase. Under normal circumstances, FGF23 has an important role in maintaining phosphorus homeostasis. In patients of TIO however, circulating level of FGF23 are high which leads to renal phosphate wasting.[2]
FGF23 also has a central role in the pathogenesis of autosomal dominant hypophosphatemic rickets (ADHR) which is caused by mutation in the FGF23 gene that makes peptides resistant to degradation by regulatory proteases. Some patients of TIO have normal FGF23 level. This lead to the discovery of other phosphatonins including secreted frizzled related protein 4 (SFRP-4). Matrix extracellular phosphoglycoprotein (MEPE) and fibroblast growth factor (FGF7) whose roles in TIO are under investigations.[6]
TIO is a rare condition. Our case demonstrated the importance of considering the diagnosis of TIO and assessing serum phosphorus levels in patients with unusual osteomalacia and persistent musculoskeletal symptoms. Measurement of serum phosphorus is an important step for understanding phosphaturia. Also with the levels of serum calcium, alkaline phosphate, PTH, 25(OH) D3, 1,25 dihydroxyvitamin D should be measured to distinguish other causes of hypophosphatemia and osteomalacia.
A spot 2 hour urine sample was collected in our patient. Calculation of TMP/ GFR and a nomogram was used to derive this from serum phosphorus and tubular reabsorption of phosphate (TRP=1- [urine phosphorus] x [serum creatinine]/[urine creatinine] x [serum phosphorus level]). This is a reliable tool for evaluation of hypophosphatemia. The patient was not on any phosphorous supplement at that time. In our patient, TMP/GFR was low despite hypophosphatemia which indicated renal phosphorus wasting.
FGF23 is a confirmatory test for diagnosis of TIO. In one survey, 22 patients with clinical evidence of TIO had elevated FGF23. Tumours associated with TIO originate from bone or soft tissue and are typically benign in nature. The common histologic type is mesenchymal tumour mixed connective tissue type, Since resection of such a tumour is a definitive treatment, localisation of a tumour is essential.[10]
In our patient, the source of the tumour was readily visible. But tumours can occur in elusive sites and their localisation can be challenging. Careful physical examination with special attention to oral cavity and extremities is needed.
For tumour localisation, imaging modalities such as X-ray, CT and MRI can be used. Some tumours express surface somatostatin receptors. Somatostatin receptor PET scan is used as a diagnostic tool in refractory cases. In our patient Somatostatin receptor PET scan was used as nuclear imaging method. Radiolabelled octreotide was injected in a patient and whole body follow images were taken. Tumours that have surface somatostatin receptors bind to the octreotide and light up images.
Renal phosphate wasting and abnormal vitamin D metabolism can also be seen in x-linked hyphosphatemia (XLH) and ADHR (Autosomal dominant hypophosphatemic rickets) and both are caused by genetic mutations. As indicated by their names, they differ in mode of inheritance. These disorders present in childhood, although ADHR can exhibit variable and delayed onset. In all these syndromes circulating phosphatonins lead to inhibition of renal tubular phosphate reabsorption and downregulation of renal 1α hydroxylase.[1]
TIO is managed by complete resection of the tumour which results in decrease in bone pain and muscle pain and healing of multiple fractures within several months. Until the tumour is located and resected, the patient requires 1-4 gm of phosphorus/day in divided dosage as well as calcitriol supplementation (1-3 gm/day). These lead to renal and gastrointestinal phosphorus reabsorption and prevent secondary hyperparathyroidism.[11] The patient had already received calcitriol as well as alphacalcidol before coming to hospital and had no symptomatic relief and her serum vitamin D level was normal, hence calcitriol or alphacalcidol was not started. Our patient was given oral phosphorus (4.5 gm/day), until she underwent resection of the tumour. Subsequent to the resection, her serum phosphorous values improved. She was given phosphorous supplementation, which was tapered gradually. Over the next few months, bone and muscle pain subsided and she was able to walk without support.
TIO is an acquired disorder of renal phosphate wasting that is associated with tumour. Patients with TIO present with hypophosphataemia with inappropriately suppressed 1,25 (OH)2 D concentration. Clinical symptoms include gradual onset of muscle weakness, fatigue and bone pain, especially for ankles, legs, hips and back. Insufficiency fractures are common and proximal muscles weakness can become severe enough for patients to require wheel chair or they become bedbound. This case highlights the importance of measuring serum phosphorus and phosphate clearance in patients with unusual musculo-skeletal pain and myopathic weakness. (68)Ga-DOTATE scan helps in localization of such a tumour and surgical resection can lead to total cure.
Ethical ConsiderationPatient consent form has been procured prior to the case report study.
Statement of AuthorshipAll authors have given approval to the final version submitted.
Author DisclosureAll the authors have declared no conflict of interest to the work carried out in this paper.
Funding SourceNone.
[1] Bringhurst F, Demay MB, Krane SM, Kronenberg HM. Disorders of bone and mineral metabolism. In: Harrison's Principles of Internal Medicine, 19th edition. New York: McGraw Hill; 2012.