Acromegaly is a rare, chronic disorder of excessive growth hormone (GH) and consequent overproduction of insulin growth factor-1 (IGF-1) from the liver. IGF-1 in large part mediates the effects of GH. Acromegaly is characterized phenotypically by progressive acral and facial disfigurement and is associated with cardiovascular, respiratory, metabolic, and gastrointestinal complications. Growth hormone secreting pituitary adenomas are the cause of acromegaly in 95% of patients, other rare causes are ectopic GH-secreting tumor, and ectopic or hypothalamic growth hormone releasing hormone-secreting tumor. Diagnosis of acromegaly is suggested by clinical features and confirmed by an elevated age- and sex-matched serum IGF-1 level and GH levels that fail to suppress to <1 ug/L after an oral glucose load. Confirmation of the source is done using Cranial MRI - pituitary protocol. Typically, after the MRI imaging, a GH-secreting pituitary adenoma will be identified and surgical resection of the tumor performed. Surgery is the treatment of choice and frequently results in biochemical remission after complete removal of the adenoma.
In rare circumstances, a pituitary adenoma on magnetic resonance imaging cannot be found; a search for an ectopic source of GH production is done. Even rarer is an acromegalic patient without an ectopic source and without an imaging evidence of pituitary adenoma. The treatment for this subset of patients is not well defined. We present our experience in dealing with such a case.
CASEOur patient is a 76-year-old female of Chinese descent who came in for initial consult in year 2000 at age 61 at another institution, due to symptoms of upper airway obstruction, i.e., choking, snoring and difficulty of breathing on lying supine. On examination, she was noted to have a enlarged hands, feet, tongue, lip, and nose; and a prominent mandible. She is a diabetic, maintained on oral hypoglycemic medications. An initial impression of acromegaly was given. Workup revealed an elevated IGF-1; and confirmed with an unsuppressed growth hormone levels on oral glucose challenge (Table 1). An MRI of the sella with contrast (pituitary protocol) revealed a normal pituitary. A CT scan of the neck, chest and abdomen done were also unremarkable.
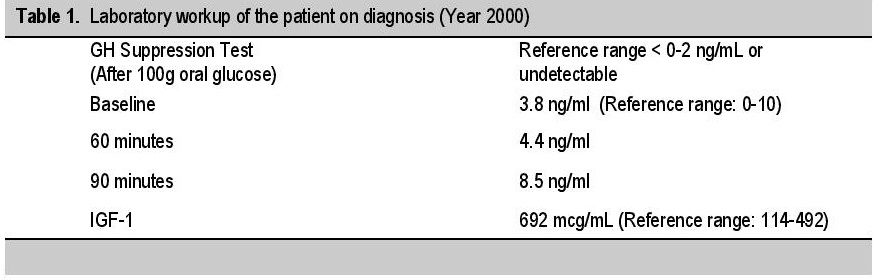
Table 1. Laboratory workup of the patient on diagnosis (Year 2000)
After explaining treatment options and the risks associated with each intervention, our patient opted for medical management. Patient was started on octreotide [Sandostatin-LAR] 20 mg monthly for 8 months, but repeat IGF-1 level was persistently elevated. Hence, cabergoline, 0.5 mg/tab, 2 tablets twice a week was added to her Sandostatin regimen. But unfortunately, due to cost, difficulty in procuring the medication, and the bothersome nature of administration (subcutaneous), patient stopped Sandostatin-LAR and continued with cabergoline, 0.5 mg, 2 tablets twice a week. A repeat pituitary MRI 2 years later (2002) showed an “empty sella.” Another MRI scan of the pituitary was done in 2004, where no pituitary adenoma was again identified (Appendix A).
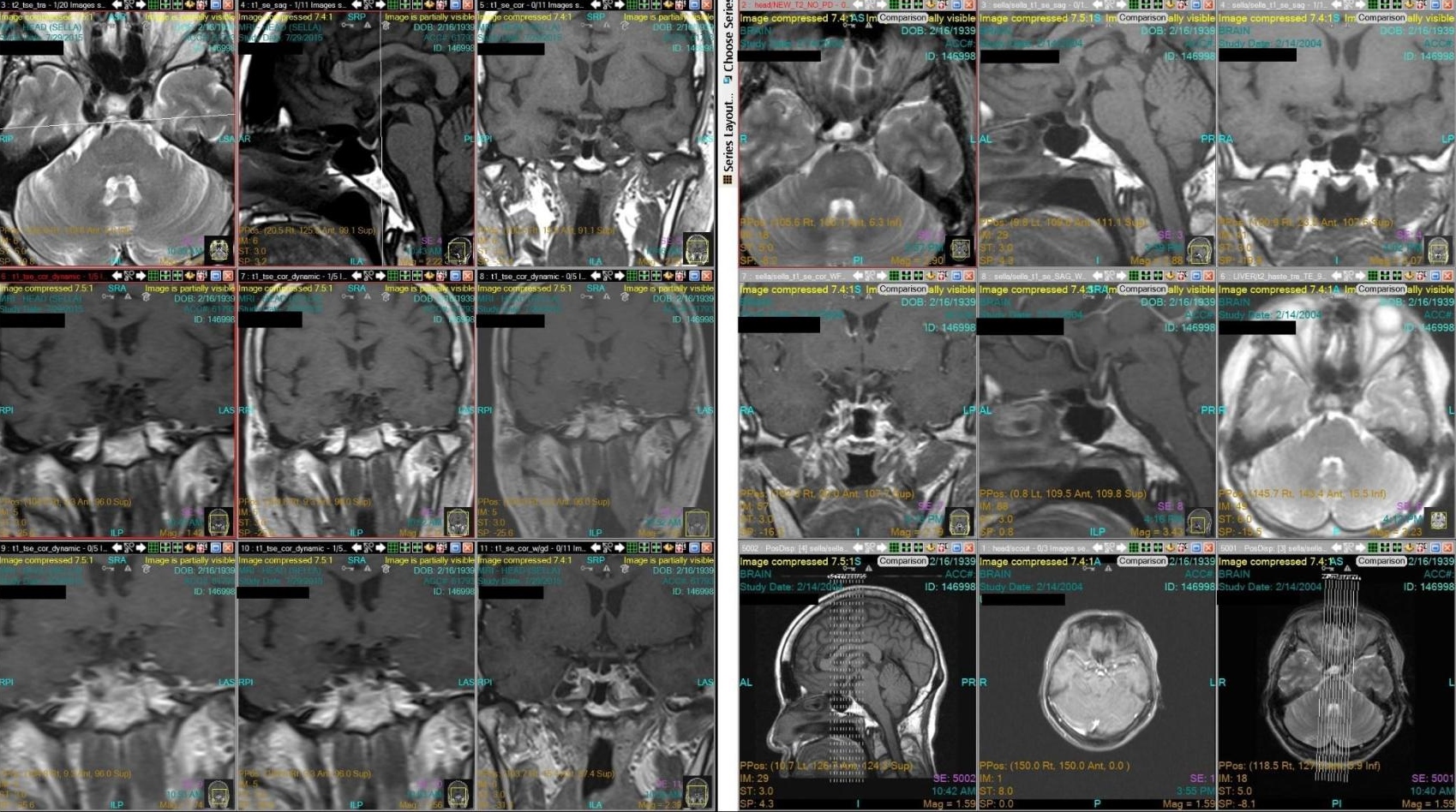
Appendix A. MRI scan of the pituitary showing pre- and post-contrast phase (left, 2015 scan; right, 2004 scan).
In June 2015, patient consulted in our institution for diabetes management follow up. The attending endocrinologist still noted coarse facial features, enlarged hands and feet (Figure 1-3). She was noted to be hypertensive on blood pressure lowering medications and diabetic on oral hypoglycemic agents. Repeat IGF-1 and levels of IGF-1 through the years are shown on Table 2. A repeat MRI imaging of the sella revealed normal pituitary, with no evidence of pituitary adenoma (Figure 4-5).
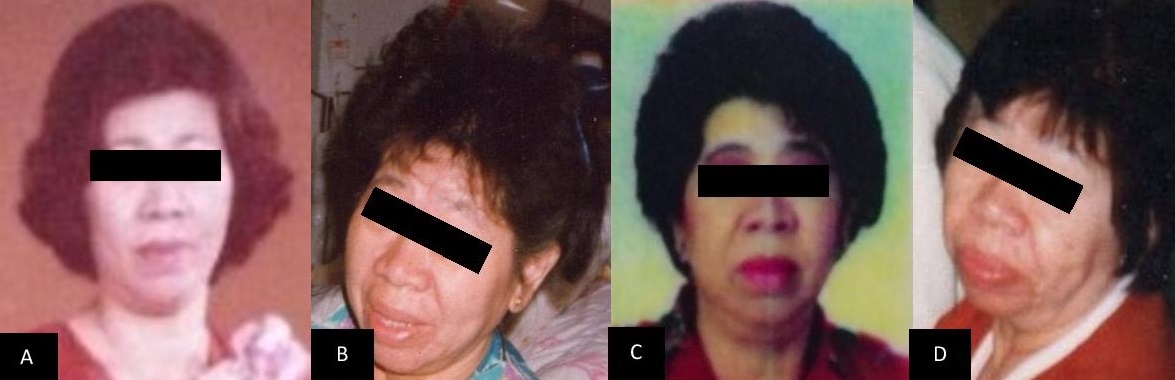
Figure 1. Pictures of the patient throughout the years (A) 1980s (B) 1989 (C) 2000 (D) 2001.
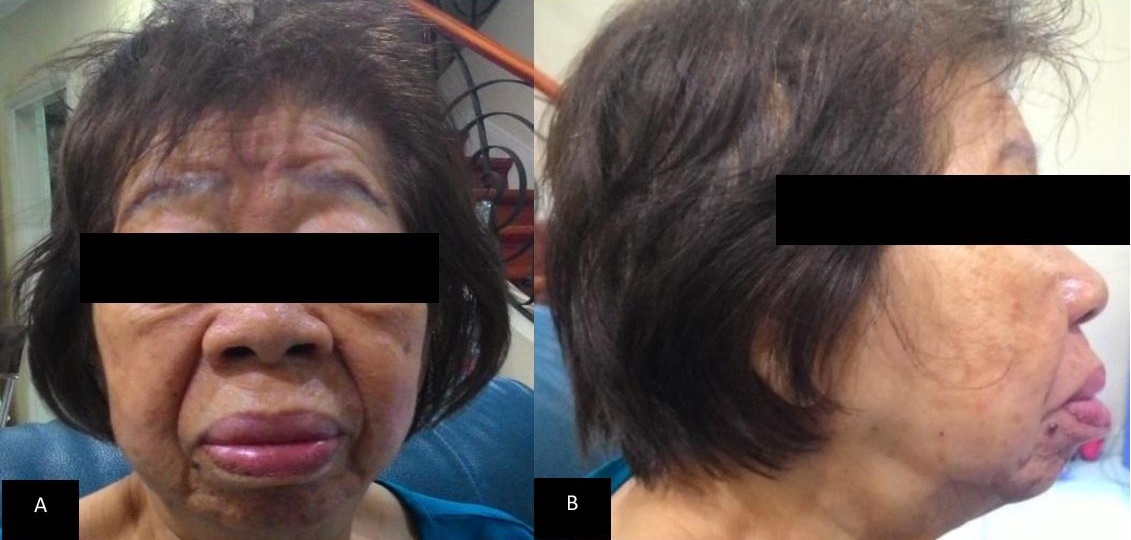
Figure 2. Latest front (a) and lateral (b) picture of the patient taken July 2015.
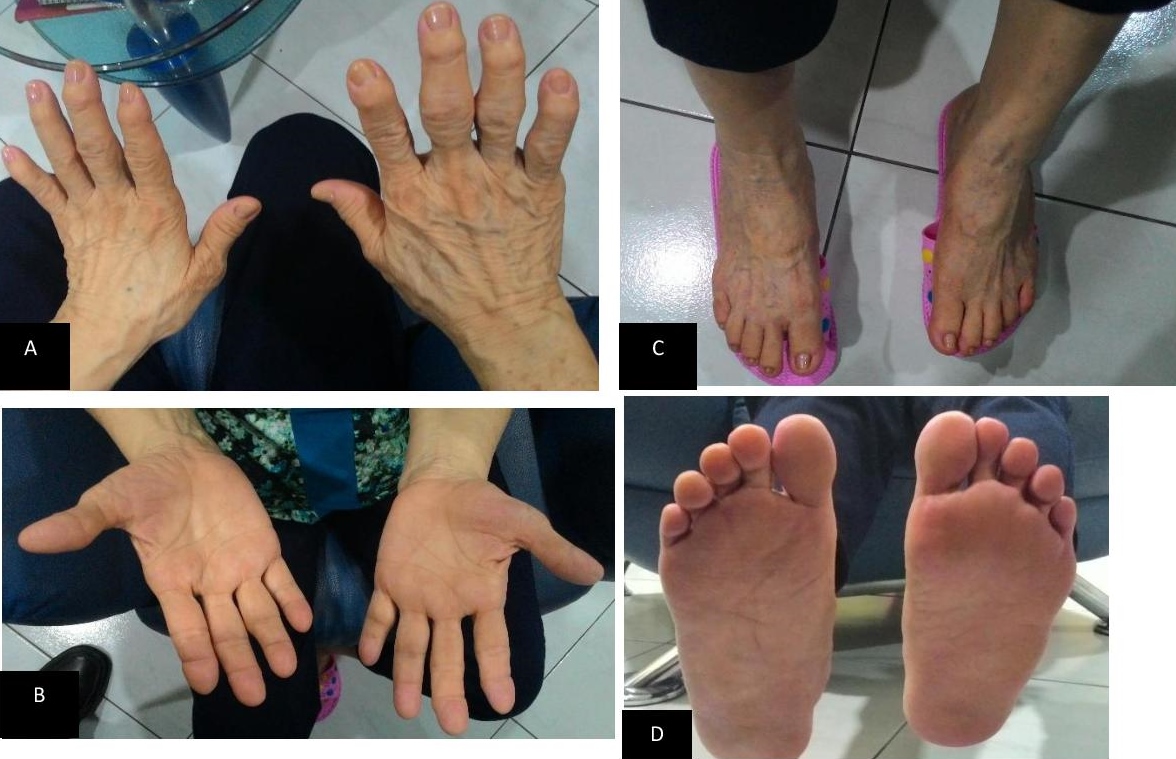
Figure 3. Picture of the patient’s hands – dorsal (A) and palms (B); patient’s feet – dorsal (C) and soles (D). Incidentally note that both her right hand and foot is larger than the left.

Table 2. IGF-1 Levels from diagnosis until present; enclosed in parenthesis are the reference values
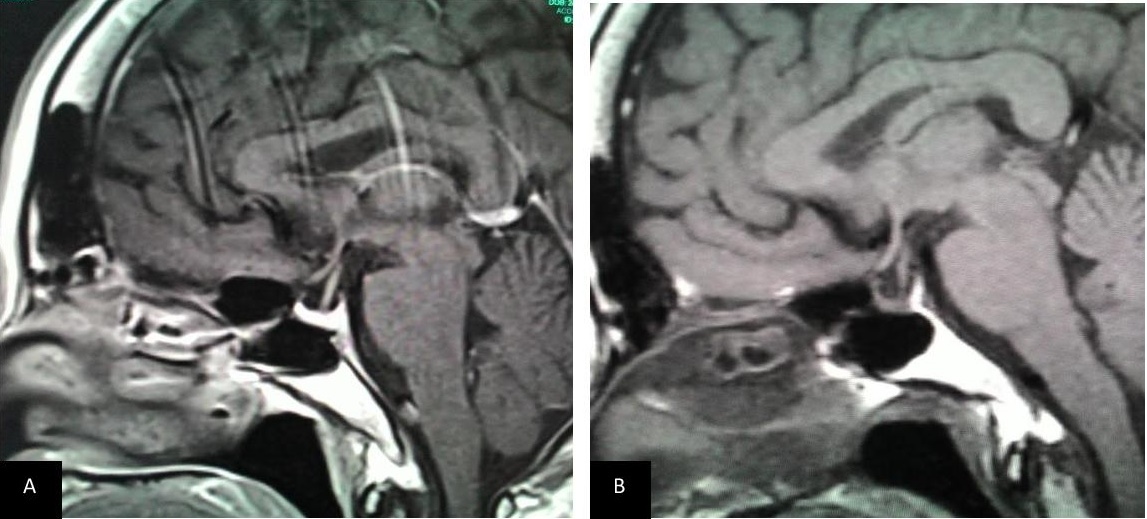
Figure 4. T1 weighted sagittal MRI image of the pituitary post contrast (A) 2015 and (B) 2004. Both showing a small pituitary gland which is pressed against the sellar floor with no internal hypoenhancement post-contrast.
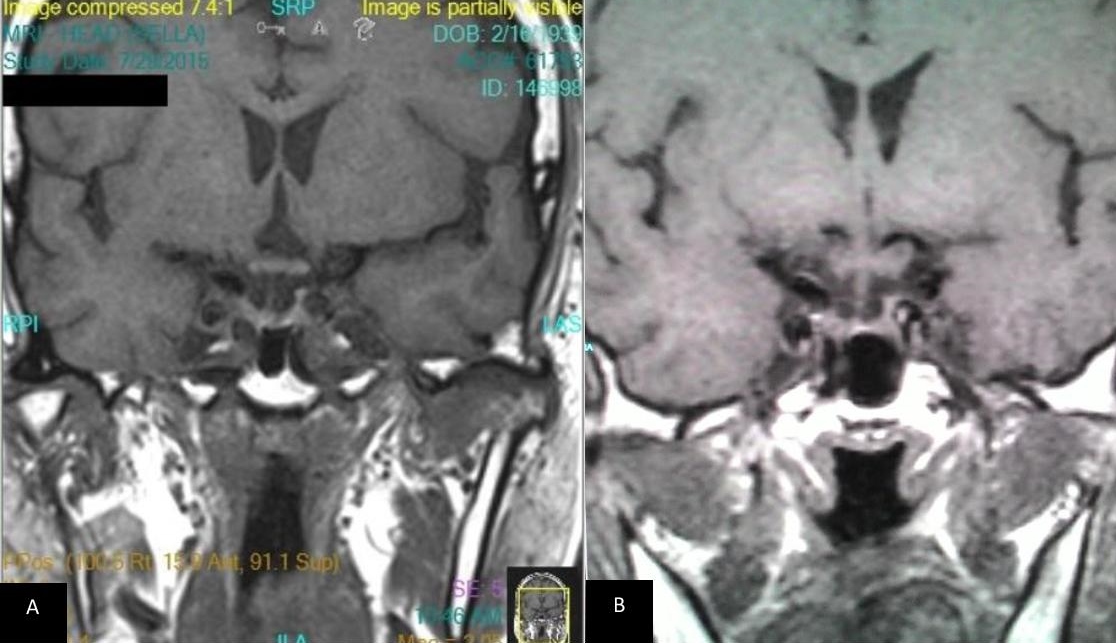
Figure 5. T1 weighted coronal MRI image of the pituitary post contrast (A) 2015 and (B) 2004. Both showing a small pituitary gland which is pressed against the sellar floor with no internal hypoenhancement post-contrast.
A hyperfunctioning growth hormone - secreting pituitary adenoma is the most common cause of acromegaly. Rarer causes are ectopic or hypothalamic growth hormone releasing hormone-secreting tumor. Since the signs and symptoms of acromegaly are indolent, the time from onset of signs and symptoms to diagnosis of acromegaly is long, usually taking years. The pituitary adenomas that cause growth hormone excess are usually large and are easily identified on routine MRI imaging of the sella. Treatment would then involve resection of the pituitary adenoma via transsphenoidal surgery.
In rare cases, a pituitary MRI may show negative results. A contrast enhanced CT scan of the chest and abdomen is the next step to detect ectopic sources of GH or GH releasing hormone (GHRH) production. Acromegaly without imaging evidence of a pituitary macroadenoma or an ectopic source is very rare. Lonser et al., reported 6 patients (mean age 56 years) with signs, symptoms and biochemical evidence of acromegaly without pituitary adenoma on imaging and an ectopic source.[1] All underwent surgical exploration of the pituitary gland and GH-secreting pituitary adenoma ranging from 5 to 6.7 mm were identified and resected. Khandelwal et al., reported one patient with acromegaly who also underwent surgical exploration of the pituitary, were a GH-secreting pituitary adenoma was identified and resected.[2]
We present our 76-year-old patient with acromegaly without pituitary evidence of a pituitary adenoma and without evidence of an ectopic source who underwent medical therapy using cabergoline 0.5 mg, 2 tablets twice a week for 14 years. Patient reports less choking and snoring symptoms and slight decrease in size of her tongue and improved sleep. Due to her age, she is not a good candidate for surgical exploration of the pituitary nor did she consent to the procedure. Other treatment options include somatostatin analogues (octreotide and lanreotide), dopamine agonists (bromocriptine and cabergoline), pegvisomant, and radiotherapy; these treatment modalities are reserved for persistent disease after surgery, for unresectable tumors or poor surgical candidates. Target goal of therapy is to reach an age-normalized serum IGF-1 value, and a random growth hormone (GH) <1.0 µg/L. A target GH <1 µg/L and normalized IGF-1 values have each been shown to correlate with mortality risk reduction.[3]
Octreotide long acting release (LAR), and deep subcutaneous lantreotide depot/autogel are administered monthly. Octreotide LAR dose is 20 mg monthly with dose titration every 3-6 months down to 10 mg or up to 40 mg monthly. Lanreotide autogel/depot starting dose is 90 mg monthly, with dose titrations down to 60 mg or up to 120 mg monthly. Serum IGF-1 and GH is measured 12 weeks just prior to the next dose. Octreotide LAR produces normalization of IGF-1 levels in only 34% of patients, but with clinical relevant reduction of GH in 72% of patients, and a significant tumor reduction in 75% of patients.[4] Lantreotide autogel, also achieves biochemical control in 34% of patients,[5] with tumor shrinkage achieved in 62.9% of patients.[6] A recently concluded phase 3 controlled trial of pasireotide, a new multireceptor targeted somatostatin analogue, reported better biochemical control and tumor size reduction in patients initially poorly responsive to first generation somatostatin analogue.[7]
Pegvisomant, a human GH receptor antagonist, competes with GH for binding at its receptor and block production of IGF-1. Pegvisomant is administered as 10, 15, or 20 mg daily injections. Serum GH levels should not be measured because GH hypersecretion persists in patients given Pegvisomant; instead, IGF-1 level is recommended as the only useful biomarker to monitor treatment efficacy. Pegvisomant reduces IGF-1 levels in 81-97% of patients,[8],[9],[10] with 5 year efficacy of 63.2% with a mean dose of 18 mg daily.[11]
Dopamine agonists (bromocriptine and cabergoline) are of limited efficacy in the treatment of acromegaly, with high doses required to achieve control. Discontinuation of treatment may result in rebound growth hormone hypersecretion. Bromocriptine is usually required at a dose of 20-30 mg per day; while cabergoline may require high doses of up to 1 mg per day to achieve control.[12] A metaanalysis showed cabergoline to be approximately 34% effective in attaining biochemical control,[13] with response appearing to decrease with time. In the study by Freda et al., in 2004, only 21% of subjects were controlled after 18 months of cabergoline administration.[14]
Sandostatin LAR administration via subcutaneous route was initially given, however, no improvement in IGF-1 levels were seen; hence, cabergoline was added to her regimen. Octreotide was eventually withdrawn due to the bothersome nature of administration and costs. She was then switched to cabergoline regimen only. Although we found no studies on the efficacy of carbergoline in the long run, the latest results of IGF-1 of our patient may indicate efficacy of cabergoline in patients with continued intake (Table 3), although further monitoring is still needed. A repeat whole abdominal CT scan done in March 2017 was still negative for an ectopic GH-producing tumor. A repeat chest and neck CT scan is still pending.

Table 3. Latest laboratory results
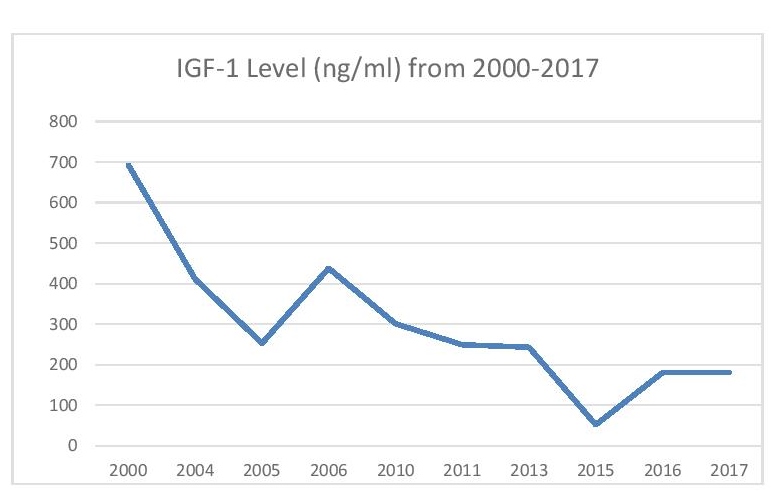
Figure 6. Line graph of IGF-1 level in ng/ml from 2000-2017.
A trial of dopamine agonist, cabergoline, may be given to patients with acromegaly and no imaging evidence of a pituitary adenoma or an ectopic source, who are poor surgical candidates and unresponsive to octreotide or other somatostatin analogues, with consideration of cost, may be given a trial of dopamine agonist, specifically cabergoline. Carbegoline therapy may have sustained response in supressing GH and IGF-1 with time, as seen in our patient who has taken cabergoline for 17 years.
Ethical ConsiderationPatient consent was obtained before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureThe authors declared no conflict of interest.
Funding SourceNone.
[1] Lonser R, Kindzelski BA, Mehta GU, Jane JA Jr., Oldfield EH. Acromegaly without imaging evidence of pituitary adenoma. J Clin Endocrinol Metab. 2010;95(9):4192-6. PubMed Central DOI.