Electrolyte imbalances, such as hyponatremia and hyperkalemia, are emergencies, especially in small infants. These abnormalities may be due to impaired mineralocorticoid secretion or response. Some causes to be considered are congenital adrenal hyperplasia (CAH) or hypoplasia, isolated aldosterone deficiency, certain medications or pseudohypoaldosteronism (PHA), a rare syndrome of mineralocorticoid resistance.[1]
PHA can be classified into Type 1 and Type 2. Type 1 PHA is a rare condition characterized by renal resistance to the actions of aldosterone and is further sub-classified into primary and secondary (transient) PHA.[2] In Type 1 primary PHA, there is mutation in the mineralocorticoid receptor that causes end organ resistance to mineralocorticoids. Type 1 secondary or transient PHA is strongly associated with urinary tract infections (UTI) complicating structural urinary tract anomalies.[2],[3]
We report a case of transient PHA in a 3-month-old baby with hyponatremic hyperkalemic metabolic acidosis to highlight that although rare, transient PHA should be considered as a differential diagnosis of hyponatremia and hyperkalemia in infants with UTI complicating structural urinary tract anomalies. Furthermore, it also points out that the existence of urinary tract infection complicating urinary tract malformations is a powerful predisposition for the development of transient aldosterone resistance.
CASEA 3-month-old, previously well Chinese girl, presented with one-week history of diarrhoea followed by one episode of afebrile seizure. She was referred from a district hospital for further work up and management. Her birth history was also unremarkable. She was delivered at term with birth weight of 3.72 kg (75th centile) and length of 55cm (97th centile) (Figure 1). She had been on exclusive breastfeeding for 1 month and then changed to infant formula. She was on multiple different brands of cow’s milk formula in view of recurrent episodes of loose stool and excessive flatulence.
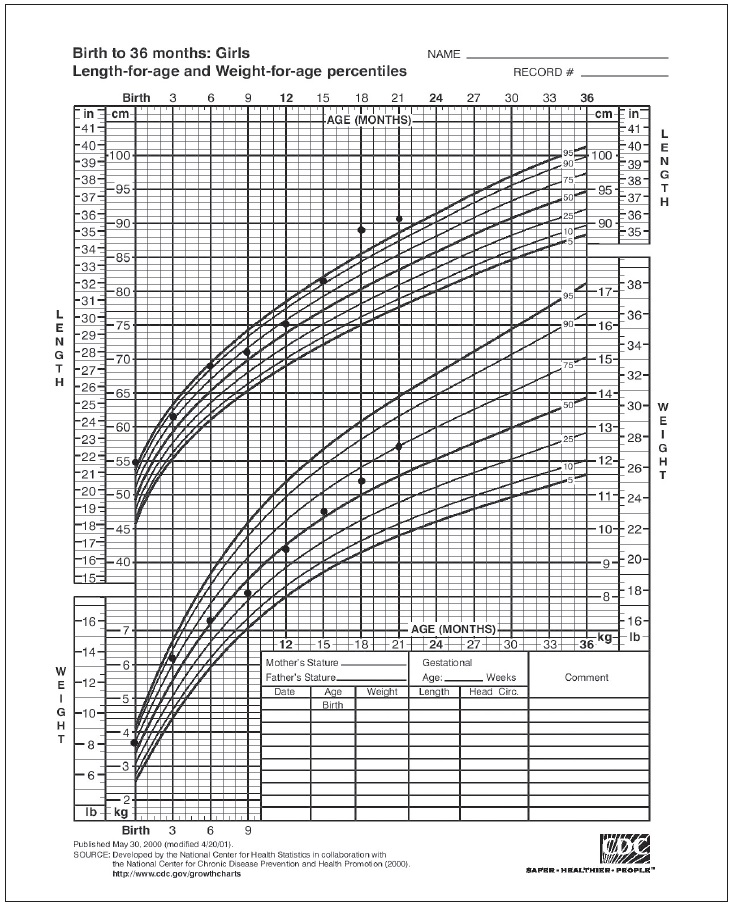
Figure 1. Growth chart from birth to 2 years old (downloaded from https://www.cdc.gov/growthcharts/clinical_charts.htm).
On examination, she was moderately dehydrated and tachycardic. Her weight was 6.2 kg (above 75th centile) and length of 61cm (75th centile). Examination revealed ballotable right kidney. Initial laboratory investigations showed hyponatremia (117 mmol/L), hypochloremia (90 mmol/l), hyperkalemia (9 mmol/L) with metabolic acidosis. She required intravenous (IV) normal saline bolus (10 ml/kg) given at the district hospital and IV hydration with 5% correction over 12 hours. She was given one cycle of lytic cocktail with calcium resonium 6 hourly to correct the hyperkalemia. Her electrolytes normalized after 4 days of treatment. Stool culture for rotavirus and reducing sugar were negative. In view of the possibility of cow’s milk protein allergy, she was commenced on extensive hydrolysed milk formula and subsequently responded well with resolution of diarrhoea. She had no recurrent episode of seizure and her neurological examination was unremarkable with normal ultrasound scan of the brain.
Her urine culture grew Enterobacter and therefore, she was treated with 7 days course of cefuroxime. Ultrasound of kidney, ureter, and bladder (USG KUB) showed right duplex kidney with gross hydronephrosis and hydroureter, while a micturating cystourethrogram (MCUG) showed grade 5 vesicoureteric reflux on the right side.
Routine screening for CAH is not done in our country. The diagnosis of transient PHA became apparent when the serum aldosterone level obtained on admission was elevated at 3700 pmol/l (normal range <1109 pmol/L) with normal 17-hydroxyprogesterone (17-OHP) level (0.8 nmol/L, reference range 1.0-14.12 nmol/L). Adrenocorticotropic hormone (ACTH) stimulation test was also normal. She was commenced on oral trimethoprim for urinary tract prophylaxis prior to discharge. She has no subsequent recurrent episode of electrolyte disturbances nor urinary tract infection as evidenced by negative urine culture post treatment. She is now two years old and currently under our follow up. Her growth is satisfactory with weight 12.4 kg (above 50th centile) and height 90 cm (90th centile). Her developmental milestones are appropriate and she is currently awaiting urethral implantation.
The primary function of aldosterone is reabsorption of sodium and water at the expense of potassium in the distal renal tubule. Deficiency of aldosterone or end organ resistance to its actions leads to hyponatremia, hyperkalemia, hypovolemia and metabolic acidosis. An elevated serum aldosterone level with hyponatremia and hyperkalemia in the absence of elevated 17-OHP level are the key findings to suggest Type 1 PHA. Type 1 PHA was first described in 1958 by Cheek and Perry.4 Since then, reports on type 1 PHA have been published and genetic analysis has recently identified two different forms of type 1 primary PHA: Renal PHA 1 or autosomal dominant-PHA and Systemic PHA 1 or autosomal recessive-PHA 1.[1],[2],[5]
On the other hand, transient PHA with obstructive uropathy was first described by Rodriguez-Soriano et al., in 1983.[6] Watanabe in his review documented that all patients were younger than 7 months and 80% of them suffered from both urinary tract malformation and associated UTI similar to our case.[7] Some cases of transient PHA in infants with UTI who had urinary tract abnormalities were previously documented.[8],[9] Schoen et al also discussed that the resolution of all hormonal and electrolyte abnormalities was followed by successful treatment of UTI in infants, which was similar to our case.[10] Development of aldosterone resistance may be predisposed by the development of UTI complicating urinary tract structural anomalies.
Our case presented with afebrile seizure and dehydration due to diarrhoea and urinary tract infection. The most likely cause of her seizure was hyponatraemia. Diarrhoea usually causes hypokalemic metabolic acidosis with varying levels of serum sodium; hyponatremic or hypernatremic or normal depending on the loss of water and sodium. However, the result of her investigations revealing hyponatremic hyperkalemic metabolic acidosis had alerted us that there might be underlying urinary and endocrine pathologies. Thus, we proceeded with further laboratory and imaging studies to obtain a final diagnosis. Normal 17 hydroxyprogesterone level and ACTH stimulation test excluded CAH. The diagnosis of type 1 PHA was strongly suggested by an elevated serum aldosterone.
However, prompt diagnosis of transient PHA may be difficult since the aldosterone assay is normally sent to a reference laboratory and takes several days to obtain the results. Measurement of urinary sodium level prior to treatment may reveal excessive urinary sodium excretion which suggests aldosterone deficiency or resistance. Patient’s presentation, laboratory studies, the existence of urinary tract anomalies and family history may help to differentiate between type 1 primary PHA from type 1 secondary, or transient PHA.
Our patient was diagnosed to have urinary tract infection with underlying gross hydronephrosis and hydroureter due to grade 5 vesicoureteric reflux on the right side and exhibited transient renal tubular resistance to aldosterone. After aggressive treatment, she has no subsequent recurrent episode of electrolyte disturbances nor urinary tract infection. We hypothesize that renal inflammation may cause transient tubular resistance to aldosterone independent of structural anomaly. The aldosterone resistance leading to salt wasting and high risk of recurrent urinary tract infection if the structural anomaly is not corrected may lead to failure to thrive in the future. However, our patient did not show any sign of failure to thrive with reasonably good growth. Treatment of transient PHA involves sodium chloride replacement, normalization of potassium level, antibiotic therapy and surgical intervention if indicated.
In conclusion, clinicians should have a high index of suspicion in diagnosing transient PHA in an infant presenting with hyponatremia, hyperkalemia and urinary tract infection with or without associated urinary tract anomalies typically if diagnosis of CAH was not found. Furthermore, in infants presenting with salt wasting, urine cultures should be obtained to investigate for underlying UTI.
AcknowledgmentsWe would like to thank the parents and the patient for giving consent to use the patient’s data.
Ethical ConsiderationPatient consent was obtained before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureAll the authors have declared no conflict of interest to the work carried out in this paper.
Funding Source
[1] Cheong HI. Pseudohypoaldosteronism Type 1. J Genet Med. 2013;10(2):81-7. CrossRef