Katrina Rodriguez-Asuncion, MD
Section of Endocrinology, Diabetes and Metabolism,
Department of Medicine, Makati Medical Center
2 Amorsolo Street, Legaspi Village, Makati, 1229, Philippines
Tel. No.: +632-888-8999 local 3240
E-mail: krodriguezmd@gmail.com
ORCiD: https://orcid.org/ 0000-0002-5219-6442
e-ISSN 2308-118x
Printed in the Philippines
Copyright © 2019 by the JAFES
Received April 22, 2019. Accepted June 20, 2019.
Published Online First: November 9, 2019.
Granulomatous hypophysitis is an extremely rare condition, with no established definitive treatment. An elderly Asian woman was diagnosed to have recurrent granulomatous hypophysitis 5 years after transsphenoidal surgery. No other intervention was done post-operatively. Since another surgery was not advisable due to the high probability of recurrence, she was started on a trial of oral glucocorticoids. After 3 months of steroid therapy, complete resolution of symptoms and sellar mass were achieved.
Keywords: granulomatous hypophysitis, autoimmune hypophysitis, glucocorticoidsGranulomatous hyphophysitis is a rare condition that can occur as a primary phenomenon or secondary to tuberculosis, syphilis, sarcoidosis, Wegener's granulomatosis and other inflammatory diseases. Clinical manifestations include hypopituitarism and local mass effects, with sudden onset of headache being the most common symptom. Some cases were initially diagnosed as pituitary adenoma because of their clinical and radiologic similarities, prompting surgery. Definitive management has not yet been established for granulomatous hypophysitis due to the limited number of cases and variable response to therapy.
A 70-year-old Asian female with good functional capacity and independence in activities of daily living presented in 2012 with frontal headache of sudden onset, characterized as non-radiating, intermittent, with a pain scale of 10/10 and aggravated by movement and sneezing. The pain was temporarily relieved by paracetamol and other non-steroidal anti-inflammatory drugs. There were no associated symptoms of blurring of vision, diplopia, visual field cuts, anosmia, change in shoe size, galactorrhea, nausea and vomiting. Upon consult, cranial magnetic resonance imaging (MRI) showed an enhancing suprasellar mass measuring 1.3 cm x 1.3 cm x 1.4 cm indenting the optic chiasm, consistent with pituitary macroadenoma (Figures 1A and B). Further biochemical evaluation showed decreased LH and a normal baseline perimetry studies (Table 1).
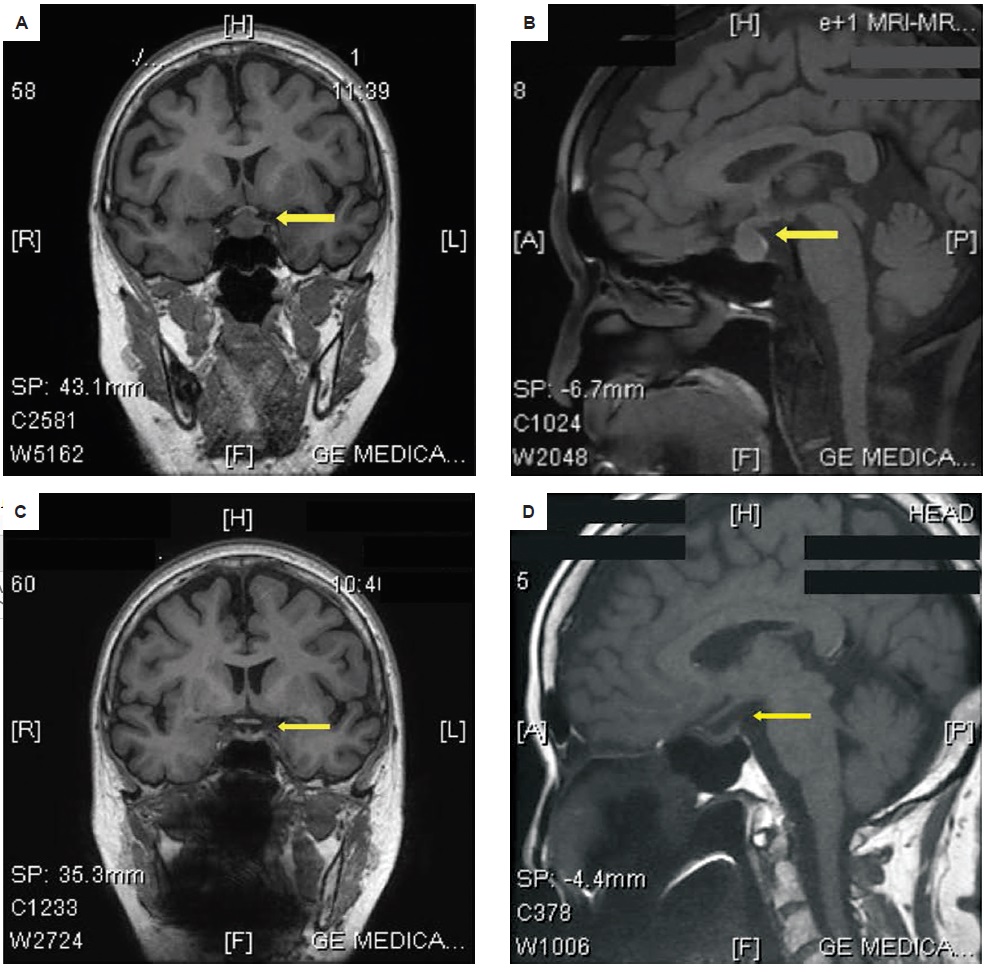
Figure 1. Preoperative cranial magnetic resonance imaging (MRI) in (A)coronal and sagittal (B) views showed an enlarged pituitary gland, with a 1.3 cm x 1.3 cm x 1.4 cm enhancing suprasellar nodule indenting the optic chiasm. Features of cystic degeneration, calcification and hemorrhage were absent. After transsphenoidal surgery, MRI showed interval resolution of the previously noted pituitary nodule, with normal appearance of the pituitary gland, optic chiasm and pituitary stalk on (C) coronal and (D) sagittal views.
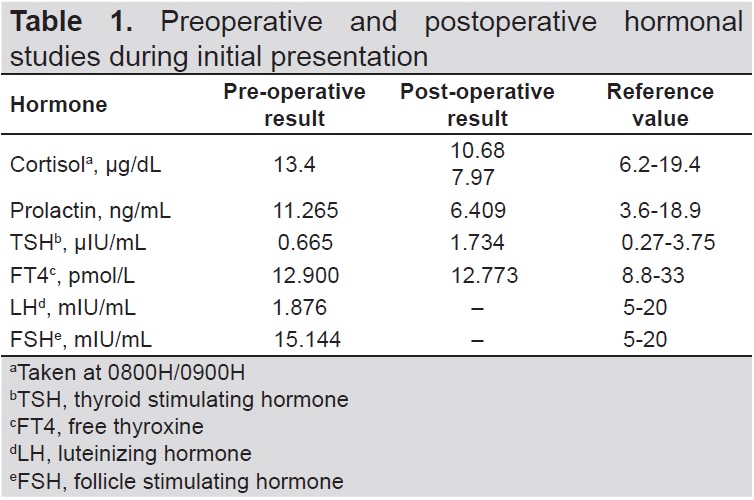
Table 1. Chest radiograph showing cardiomegaly and pulmonary congestion upon admission.
Her other comorbidities were hypertension for more than 10 years, type 2 diabetes mellitus for 10 years and osteoporosis diagnosed one year ago. She had undergone right mastectomy for ductal carcinoma in situ 18 years ago, with no subsequent radiation or chemotherapy. Her menstrual history was unremarkable. She was nulligravid despite normal fertility work up and clomiphene intake for almost 3 years. She had also undergone cataract surgery and laparoscopic cholecystectomy. Her medications were sitagliptin, metformin, amlodipine, losartan, tamoxifen and ibandronic acid. She had no history of steroid use.
The clinical impression in 2012 was pituitary macroadenoma. The patient underwent transsphenoidal surgery with an unremarkable post-operative course. Post-operative cranial MRI showed interval resolution of the previously noted pituitary mass, with normal appearance of the pituitary gland including the optic chiasm and pituitary stalk (Figures 1C and 1D). Postoperative pituitary hormones showed normal results (Table 1).
The histologic features were consistent with chronic granulomatous inflammation with focal necrosis instead of a pituitary adenoma (Figure 2). Acid-fast bacilli and silver sethenamine stains showed negative results. Upon consult with another institution abroad, the diagnosis was non-caseating granulomatous inflammation involving the pituitary gland, with primary considerations of tuberculosis, giant cell granuloma (idiopathic granulomatous hypophysitis) or sarcoidosis. Mycobacterium tuberculosis PCR/nucleic acid amplification were also negative. There was no treatment rendered during that time. She was advised to have annual MRI for monitoring but the patient was lost to follow-up.
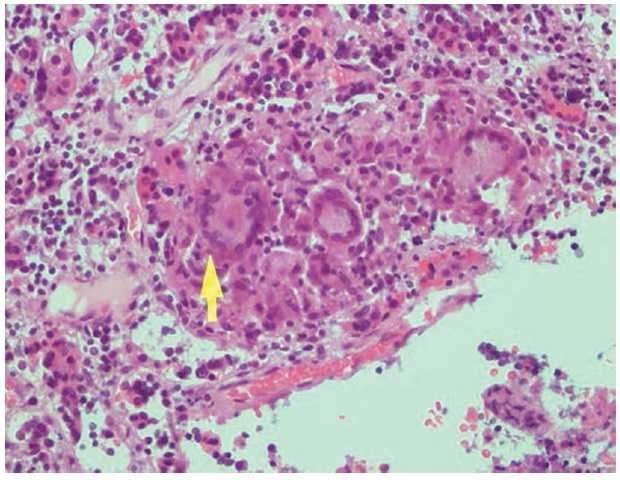
Figure 2. Photomicrograph showing granuloma formation characterized by central epithelioid, foamy macrophages and giant cells (H&E, 200x).
Interval history was unremarkable until five years after the surgery. The patient began to experience intermittent, non-radiating headache, with a pain scale of 3 to 10/10, temporarily relieved by paracetamol and tramadol. The persistence of her symptoms prompted consult with a Neurologist. Cranial MRI showed a 1.2 cm x 1.3 cm x 1.5 cm enhancing left sellar nodule (Figure 3). No visual field defects were noted on perimetry. Hormonal tests showed central hypothyroidism and hypogonadism (Table 2).
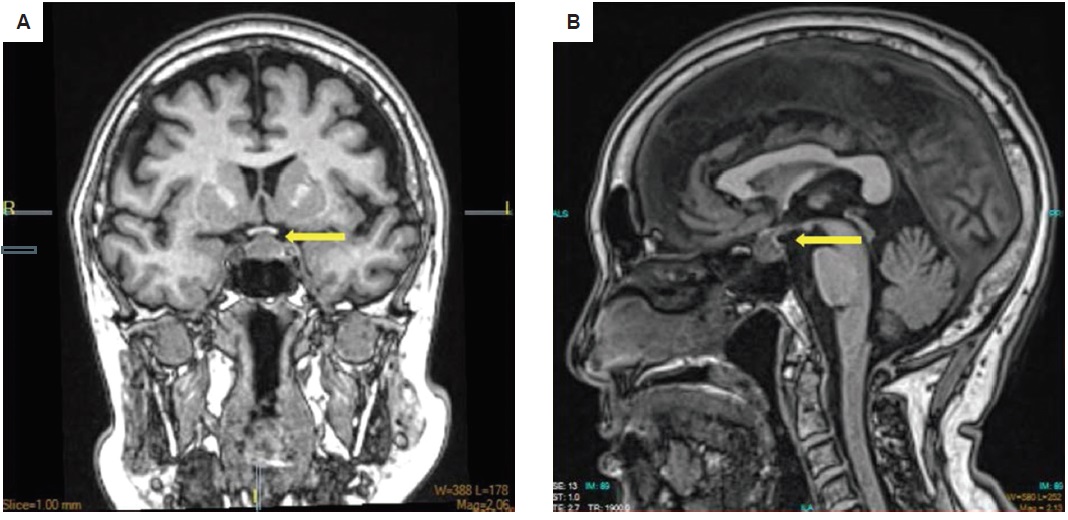
Figure 3. Cranial MRI upon recurrence of symptoms in (A) coronal and (B) sagittal views showing an enhancing left sellar nodule measuring 1.2 cm x 1.3 cm x 1.5 cm superiorly deviating the pituitary infundibulum to the right and abutting the optic chiasm.
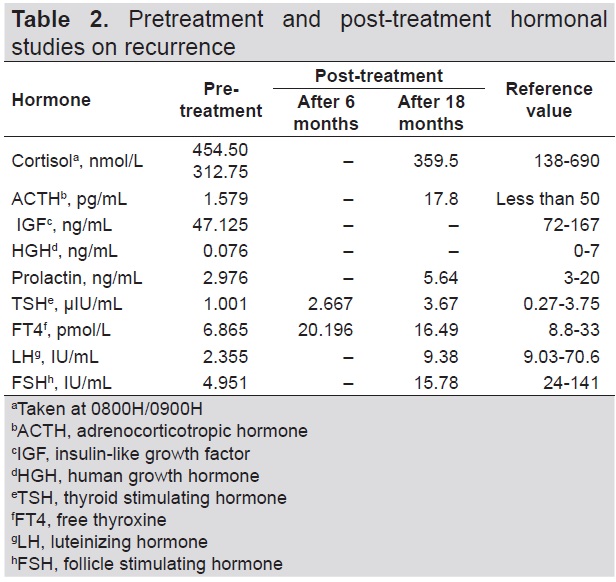
Table 2.Pretreatment and post-treatment hormonal studies on recurrence
Surgical intervention was not advised at that time due to the high probability of recurrence and eventual impairment of pituitary function. As several case reports have shown that granulomatous hypophysitis may respond to steroid therapy, the patient was started on prednisone at 30 mg per day. Resolution of symptoms were noted after initiation of treatment. Prednisone was eventually tapered by 5 mg per day every 2 weeks. After attaining a dose of 5 mg per day for two weeks, it was then given at 3 times a week, twice a week, once a week, and then discontinued, with an interval of 2 weeks each. After 3 months of steroid therapy, cranial MRI showed complete resolution of the sellar mass (Figures 4A and 4B). During the course of treatment, the patient was clinically evaluated for the possible side effects of the steroid therapy and symptomatic hypothyroidism. Aside from the weight gain which completely resolved after treatment, there were no other adverse effects noted, particularly hypotension, uncontrolled hypertension, hypoglycemia, uncontrolled hyperglycemia, headache, body weakness, constipation and electrolyte imbalance. Upon reevaluation approximately 6 months post-treatment, cranial MRI showed no enhancing lesion in the pituitary gland (Figures 4C and 4D) and normal thyroid function (Table 2).
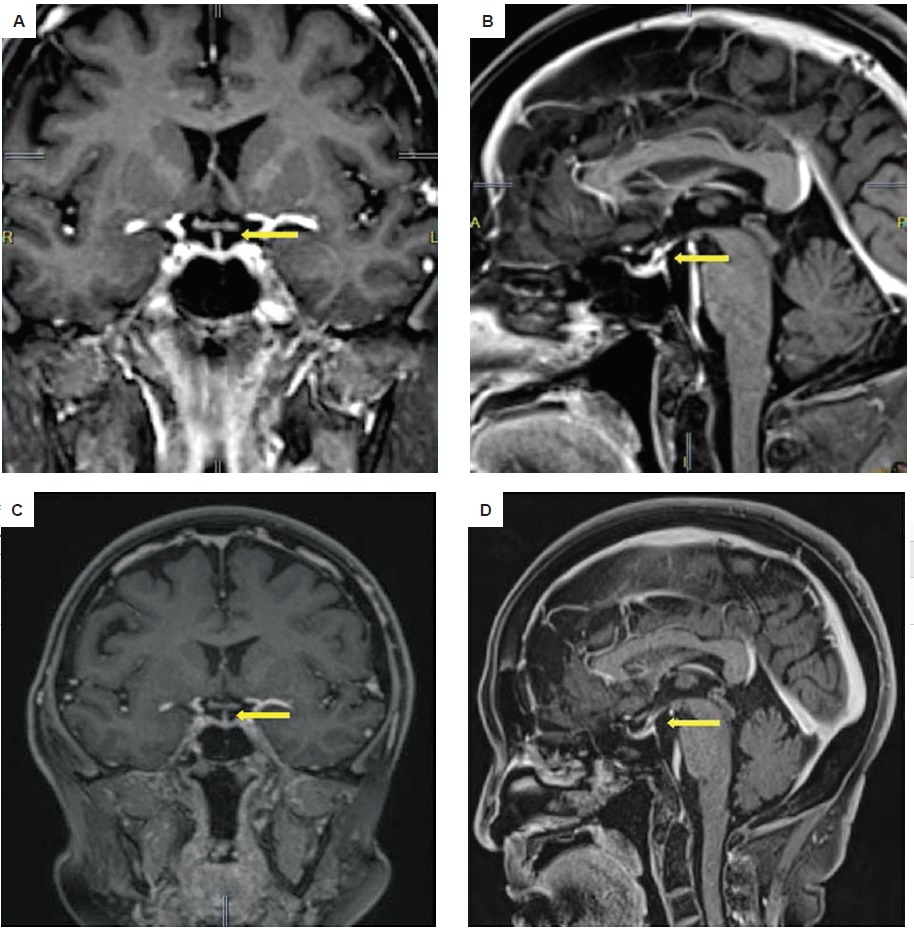
Figure 4. Cranial MRI in (A) coronal and (B) sagittal views after 3 months of steroid therapy showing complete resolution of the sellar nodule. A normal-sized pituitary gland, midline infundibulum and normal optic chiasm on both views (C and D) were seen approximately 6 months after treatment.
Hypophysitis is a rare inflammatory disorder of the pituitary gland, which can be classified as lymphocytic, granulomatous, xanthomatous, xanthogranulomatous or necrotizing.[1] Granulomatous hypophysitis, one of its most common types, has an estimated incidence of one case per 9 million people per year.[2] This condition is mostly seen in females especially in older age groups.
Granulomatous hypophysitis can also be caused by systemic diseases characterized by granuloma formation such as sarcoidosis, syphilis and tuberculosis. It is also associated with other intrasellar lesions such as adenomas, mucoceles and Rathke’s cleft cysts.[1],[2]
Signs and symptoms of granulomatous hypophysitis, particularly visual disturbances, headaches and hypopituitarism, are usually secondary to mass effect. In a systematic review of 82 cases of idiopathic granulomatous hypophysitis, the most common presenting symptoms were headache and visual disturbances.[3]
Biochemical findings appear to be normal in some cases. This may be attributed to a non-functioning pituitary adenoma, as seen in our patient during the first occurrence of the mass. Abnormal results were frequently associated with low adrenocorticotropic hormone and/or cortisol, low growth hormone, central hypothyroidism, and low follicle stimulating hormone (FSH) or luteinizing hormone (LH).[3] Low to low normal free thyroxine, thyroid stimulating hormone, FSH and LH were seen in our patient during recurrence of the disease, most probably secondary to mass effect.
Granulomatous hypophysitis is usually diagnosed on the basis of histopathologic findings of presence of granuloma formation, multinucleated giant cells, plasma cells, and/or lymphocytes. Computed tomography and MRI are unable to differentiate hypophysitis from pituitary adenoma, as these show similar degrees of enhancement. Granulomatous hypophysitis appears to be homogeneously iso-intense to brain parenchyma on MRI.[4]
Due to its rarity, definitive treatment for granulomatous hypophysitis has not yet been established. A descriptive analysis of different treatment modalities (excision, excision and corticosteroid, and biopsy and corticosteroid) showed a higher recurrence rate in individuals who underwent excision and corticosteroid, compared to biopsy with steroid.[3] It has been suggested that these conditions should be managed conservatively. Transsphenoidal surgery should only be considered when the patient presents with visual symptoms, with the procedure having both diagnostic and therapeutic purpose. Steroid therapy can be initiated postoperatively and maintained for a period of time.[5]
Granulomatous hypophysitis is a rare chronic inflammatory disorder of the pituitary gland that may present similar to a pituitary adenoma. The diagnosis can only be made histopathologically. The natural course of granulomatous hypophysitis is not completely understood. As an inflammatory condition, most cases responded well with high dose steroid therapy. In our patient’s case, glucocorticoid treatment in tapering doses showed complete resolution of the symptoms and the pituitary lesion, with no noted detrimental effects of steroid therapy. This treatment spared the patient from surgery that may have conferred deleterious consequences. Due to the rarity of this condition as well as its variable response to treatment, long-term follow-up is warranted to detect recurrence early.
Ethical ConsiderationPatient consent was obtained before submission of the manuscript.
Statement of AuthorshipAll authors certified fulfillment of ICMJE authorship criteria.
Author DisclosureThe authors declared no conflict of interest
Funding SourceNone.
[1] Prete A, Salvatori R. Hypophysitis. 2018. In: Feingold KR, Anawalt
B, Boyce A, et al., eds. Endotext [Internet]. South Dartmouth, MA:
MDText.com, Inc., 2000-.
https://www.ncbi.nlm.nih.gov/books/ NBK519842/.
[2] Park HJ, Park SH, Kim JH, Kim YH. Idiopathic Granulomatous
Hypophysitis with Rapid Onset: A Case Report. Brain Tumor Res
Treat. 2019 Apr;7(1):57-61.
[3] Hunn BHM, Martin WG, Simpson S, Mclean CA. Idiopathic
granulomatous hypophysitis: a systematic review of 82 cases in the
literature. Pituitary. 2014;17(4):357-65.
[4] Elgamal ME, Mohamed RMH, Fiad T, Elgamal EA. Granulomatous
hypophysitis: rare disease with challenging diagnosis. Clin Case Rep.
2017;5(7):1147-51.
PubMed PubMed Central CrossRef
[5] Shi J, Zhang J, Wu Q, Chen G, Zhang H, Bo W. Granulomatous
hypophysitis: two case reports and literature review. J Zhejiang Univ
Sci B. 2009;10(7):552-8.
PubMed PubMed Central CrossRef
Authors are required to accomplish, sign and submit scanned copies of the JAFES Author Form consisting of: (1) Authorship Certification, that all the requirements for authorship have been met by each author, and that the final version of the manuscript has been read and approved by all authors; (2) the Author Declaration, that the article represents original material that is not being considered for publication or has not been published or accepted for publication elsewhere; (3) the Statement of Copyright Transfer [accepted manuscripts become the permanent property of the JAFES and are licensed with an Attribution-Share Alike-Non-Commercial Creative Commons License. Articles may be shared and adapted for non-commercial purposes as long as they are properly cited]; and the ICMJE form for Disclosure of Potential Conflicts of Interest. For original articles, authors are required to submit a scanned copy of the Ethics Review Approval of their research as well as registration in trial registries as appropriate. For manuscripts reporting data from studies involving animals, authors are required to submit a scanned copy of the Institutional Animal Care and Use Committee approval. For Case Reports or Series, and Images in Endocrinology, consent forms, are required for the publication of information about patients; otherwise, appropriate ethical clearance has been obtained from the institutional review board. Articles and any other material published in the JAFES represent the work of the author(s) and should not be construed to reflect the opinions of the Editors or the Publisher.